 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards a Modular Architecture for Science Factories
Aug 18, 2023Advances in robotic automation, high-performance computing (HPC), and artificial intelligence (AI) encourage us to conceive of science factories: large, general-purpose computation- and AI-enabled self-driving laboratories (SDLs) with the generality and scale needed both to tackle large discovery problems and to support thousands of scientists. Science factories require modular hardware and software that can be replicated for scale and (re)configured to support many applications. To this end, we propose a prototype modular science factory architecture in which reconfigurable modules encapsulating scientific instruments are linked with manipulators to form workcells, that can themselves be combined to form larger assemblages, and linked with distributed computing for simulation, AI model training and inference, and related tasks. Workflows that perform sets of actions on modules can be specified, and various applications, comprising workflows plus associated computational and data manipulation steps, can be run concurrently. We report on our experiences prototyping this architecture and applying it in experiments involving 15 different robotic apparatus, five applications (one in education, two in biology, two in materials), and a variety of workflows, across four laboratories. We describe the reuse of modules, workcells, and workflows in different applications, the migration of applications between workcells, and the use of digital twins, and suggest directions for future work aimed at yet more generality and scalability. Code and data are available at https://ad-sdl.github.io/wei2023 and in the Supplementary Information
Pandemic Drugs at Pandemic Speed: Accelerating COVID-19 Drug Discovery with Hybrid Machine Learning- and Physics-based Simulations on High Performance Computers
Mar 04, 2021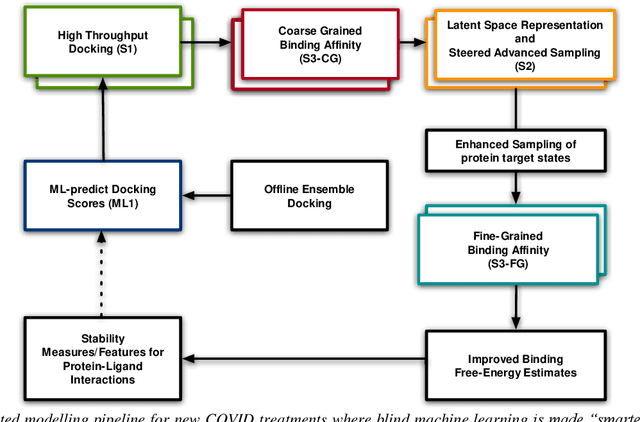

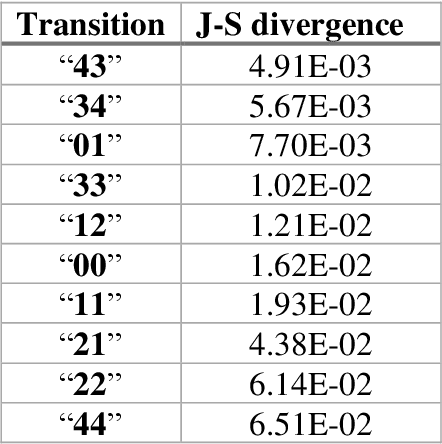
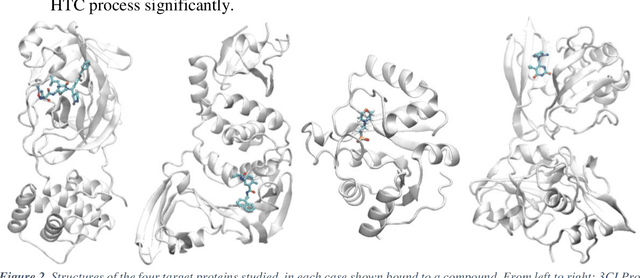
The race to meet the challenges of the global pandemic has served as a reminder that the existing drug discovery process is expensive, inefficient and slow. There is a major bottleneck screening the vast number of potential small molecules to shortlist lead compounds for antiviral drug development. New opportunities to accelerate drug discovery lie at the interface between machine learning methods, in this case developed for linear accelerators, and physics-based methods. The two in silico methods, each have their own advantages and limitations which, interestingly, complement each other. Here, we present an innovative method that combines both approaches to accelerate drug discovery. The scale of the resulting workflow is such that it is dependent on high performance computing. We have demonstrated the applicability of this workflow on four COVID-19 target proteins and our ability to perform the required large-scale calculations to identify lead compounds on a variety of supercomputers.
Targeting SARS-CoV-2 with AI- and HPC-enabled Lead Generation: A First Data Release
May 28, 2020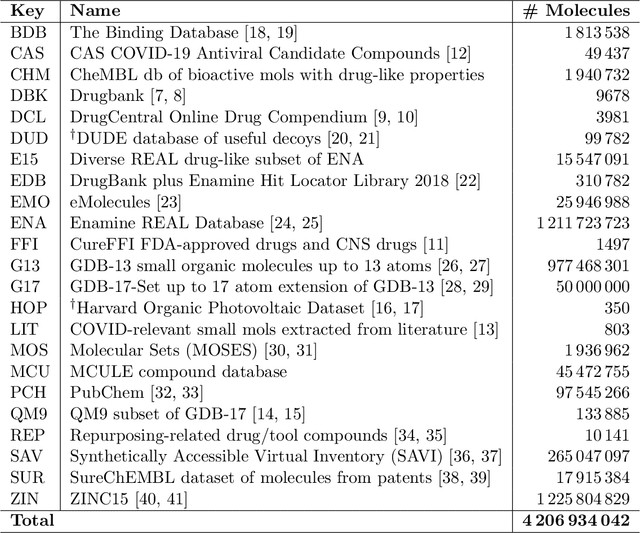
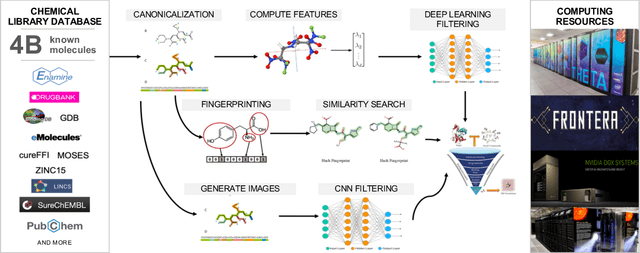

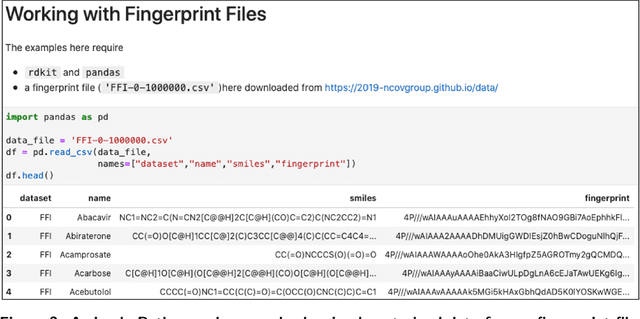
Researchers across the globe are seeking to rapidly repurpose existing drugs or discover new drugs to counter the the novel coronavirus disease (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). One promising approach is to train machine learning (ML) and artificial intelligence (AI) tools to screen large numbers of small molecules. As a contribution to that effort, we are aggregating numerous small molecules from a variety of sources, using high-performance computing (HPC) to computer diverse properties of those molecules, using the computed properties to train ML/AI models, and then using the resulting models for screening. In this first data release, we make available 23 datasets collected from community sources representing over 4.2 B molecules enriched with pre-computed: 1) molecular fingerprints to aid similarity searches, 2) 2D images of molecules to enable exploration and application of image-based deep learning methods, and 3) 2D and 3D molecular descriptors to speed development of machine learning models. This data release encompasses structural information on the 4.2 B molecules and 60 TB of pre-computed data. Future releases will expand the data to include more detailed molecular simulations, computed models, and other products.
A Systematic Approach to Featurization for Cancer Drug Sensitivity Predictions with Deep Learning
May 04, 2020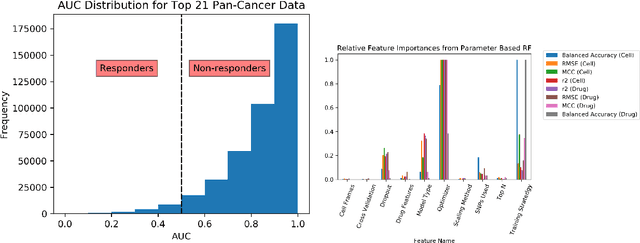
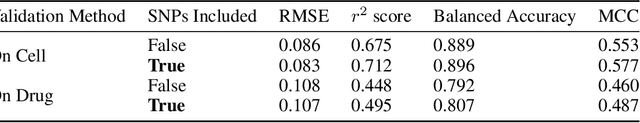

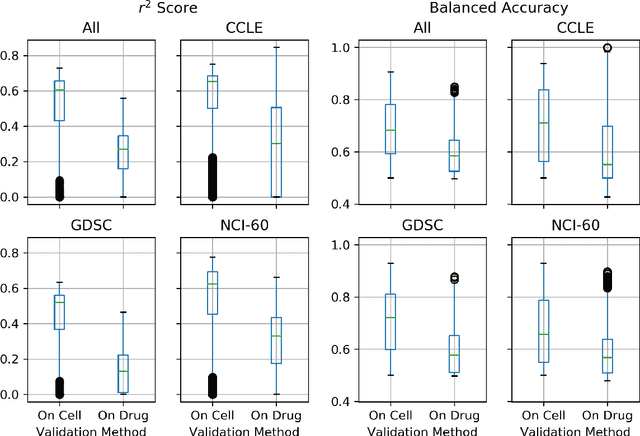
By combining various cancer cell line (CCL) drug screening panels, the size of the data has grown significantly to begin understanding how advances in deep learning can advance drug response predictions. In this paper we train >35,000 neural network models, sweeping over common featurization techniques. We found the RNA-seq to be highly redundant and informative even with subsets larger than 128 features. We found the inclusion of single nucleotide polymorphisms (SNPs) coded as count matrices improved model performance significantly, and no substantial difference in model performance with respect to molecular featurization between the common open source MOrdred descriptors and Dragon7 descriptors. Alongside this analysis, we outline data integration between CCL screening datasets and present evidence that new metrics and imbalanced data techniques, as well as advances in data standardization, need to be developed.
Scalable Reinforcement-Learning-Based Neural Architecture Search for Cancer Deep Learning Research
Sep 01, 2019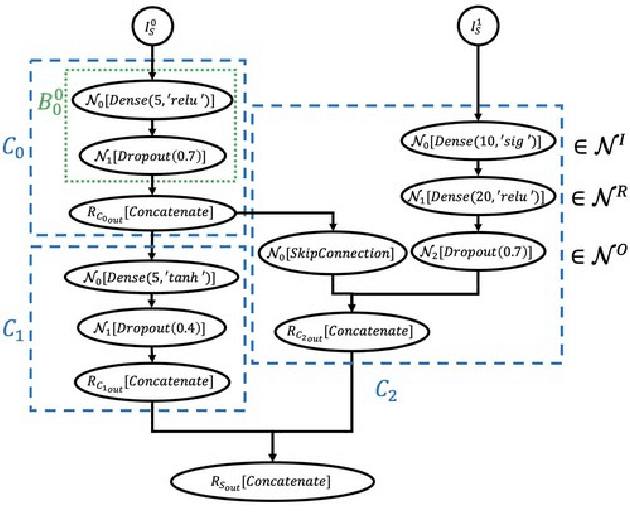
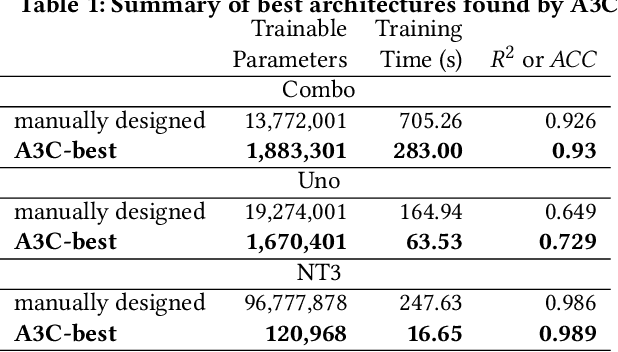
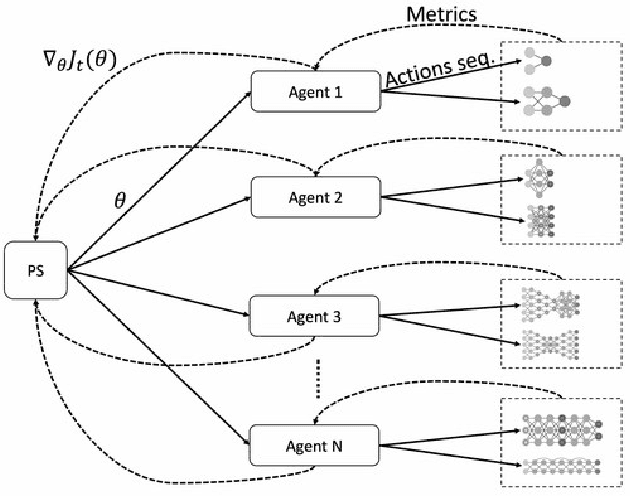
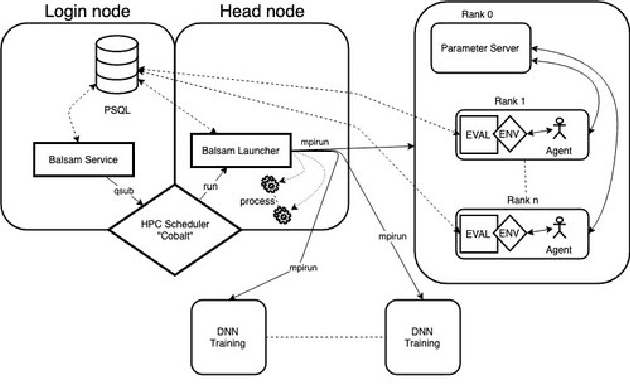
Cancer is a complex disease, the understanding and treatment of which are being aided through increases in the volume of collected data and in the scale of deployed computing power. Consequently, there is a growing need for the development of data-driven and, in particular, deep learning methods for various tasks such as cancer diagnosis, detection, prognosis, and prediction. Despite recent successes, however, designing high-performing deep learning models for nonimage and nontext cancer data is a time-consuming, trial-and-error, manual task that requires both cancer domain and deep learning expertise. To that end, we develop a reinforcement-learning-based neural architecture search to automate deep-learning-based predictive model development for a class of representative cancer data. We develop custom building blocks that allow domain experts to incorporate the cancer-data-specific characteristics. We show that our approach discovers deep neural network architectures that have significantly fewer trainable parameters, shorter training time, and accuracy similar to or higher than those of manually designed architectures. We study and demonstrate the scalability of our approach on up to 1,024 Intel Knights Landing nodes of the Theta supercomputer at the Argonne Leadership Computing Facility.