 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtein-Ligand Docking Surrogate Models: A SARS-CoV-2 Benchmark for Deep Learning Accelerated Virtual Screening
Jun 30, 2021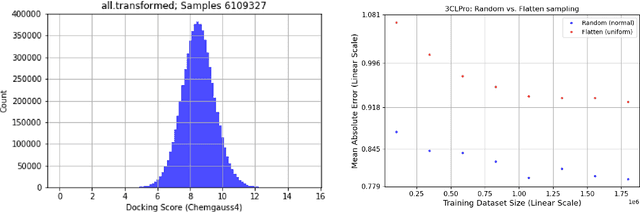

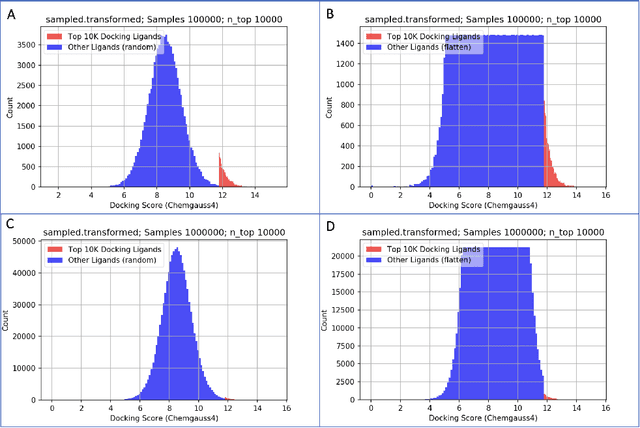
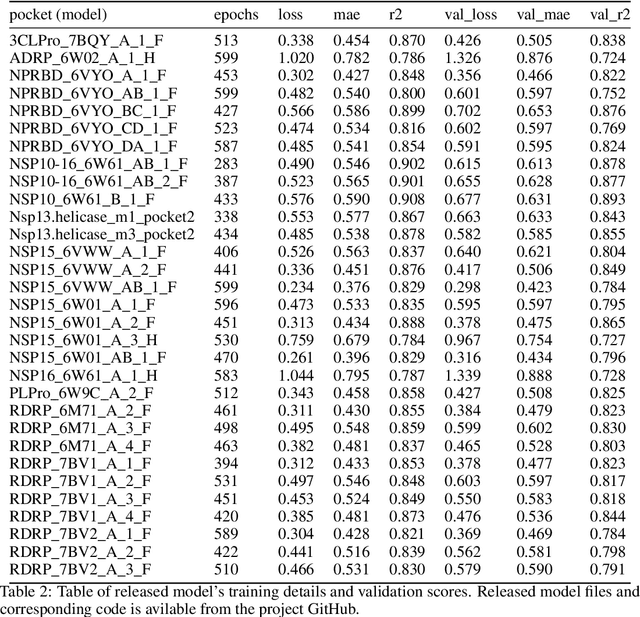
We propose a benchmark to study surrogate model accuracy for protein-ligand docking. We share a dataset consisting of 200 million 3D complex structures and 2D structure scores across a consistent set of 13 million "in-stock" molecules over 15 receptors, or binding sites, across the SARS-CoV-2 proteome. Our work shows surrogate docking models have six orders of magnitude more throughput than standard docking protocols on the same supercomputer node types. We demonstrate the power of high-speed surrogate models by running each target against 1 billion molecules in under a day (50k predictions per GPU seconds). We showcase a workflow for docking utilizing surrogate ML models as a pre-filter. Our workflow is ten times faster at screening a library of compounds than the standard technique, with an error rate less than 0.01\% of detecting the underlying best scoring 0.1\% of compounds. Our analysis of the speedup explains that to screen more molecules under a docking paradigm, another order of magnitude speedup must come from model accuracy rather than computing speed (which, if increased, will not anymore alter our throughput to screen molecules). We believe this is strong evidence for the community to begin focusing on improving the accuracy of surrogate models to improve the ability to screen massive compound libraries 100x or even 1000x faster than current techniques.
Bridge Data Center AI Systems with Edge Computing for Actionable Information Retrieval
May 28, 2021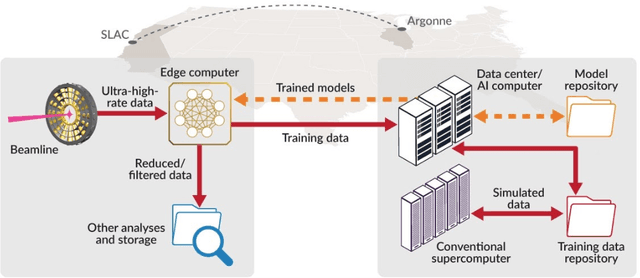

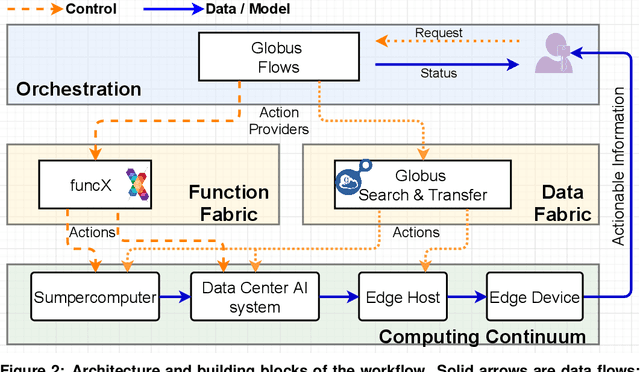
Extremely high data rates at modern synchrotron and X-ray free-electron lasers (XFELs) light source beamlines motivate the use of machine learning methods for data reduction, feature detection, and other purposes. Regardless of the application, the basic concept is the same: data collected in early stages of an experiment, data from past similar experiments, and/or data simulated for the upcoming experiment are used to train machine learning models that, in effect, learn specific characteristics of those data; these models are then used to process subsequent data more efficiently than would general-purpose models that lack knowledge of the specific dataset or data class. Thus, a key challenge is to be able to train models with sufficient rapidity that they can be deployed and used within useful timescales. We describe here how specialized data center AI systems can be used for this purpose.
Learning Curves for Drug Response Prediction in Cancer Cell Lines
Nov 25, 2020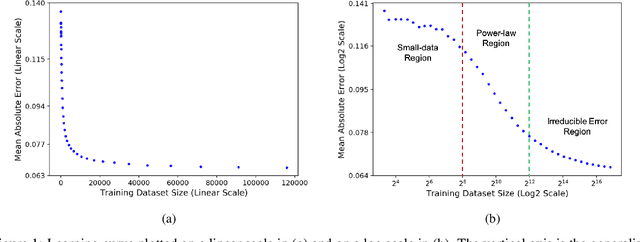
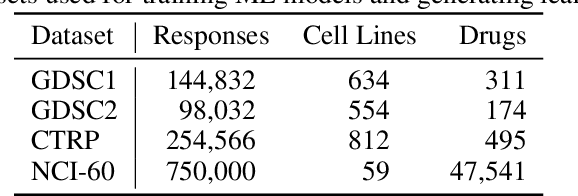
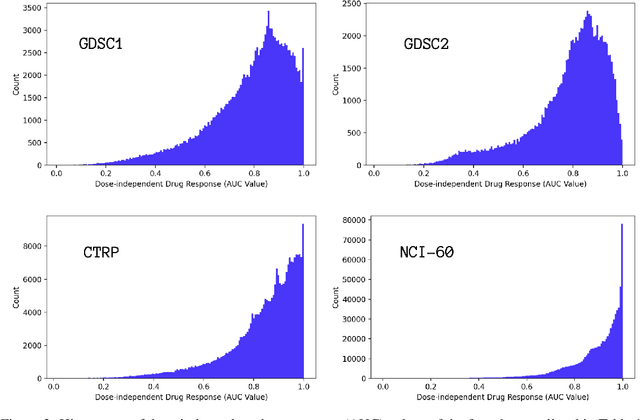
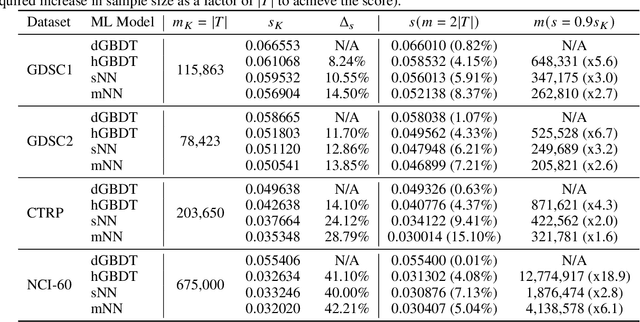
Motivated by the size of cell line drug sensitivity data, researchers have been developing machine learning (ML) models for predicting drug response to advance cancer treatment. As drug sensitivity studies continue generating data, a common question is whether the proposed predictors can further improve the generalization performance with more training data. We utilize empirical learning curves for evaluating and comparing the data scaling properties of two neural networks (NNs) and two gradient boosting decision tree (GBDT) models trained on four drug screening datasets. The learning curves are accurately fitted to a power law model, providing a framework for assessing the data scaling behavior of these predictors. The curves demonstrate that no single model dominates in terms of prediction performance across all datasets and training sizes, suggesting that the shape of these curves depends on the unique model-dataset pair. The multi-input NN (mNN), in which gene expressions and molecular drug descriptors are input into separate subnetworks, outperforms a single-input NN (sNN), where the cell and drug features are concatenated for the input layer. In contrast, a GBDT with hyperparameter tuning exhibits superior performance as compared with both NNs at the lower range of training sizes for two of the datasets, whereas the mNN performs better at the higher range of training sizes. Moreover, the trajectory of the curves suggests that increasing the sample size is expected to further improve prediction scores of both NNs. These observations demonstrate the benefit of using learning curves to evaluate predictors, providing a broader perspective on the overall data scaling characteristics. The fitted power law curves provide a forward-looking performance metric and can serve as a co-design tool to guide experimental biologists and computational scientists in the design of future experiments.
Ensemble Transfer Learning for the Prediction of Anti-Cancer Drug Response
May 13, 2020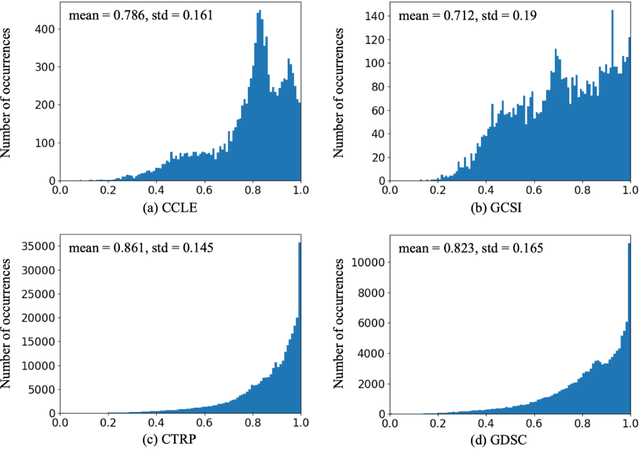
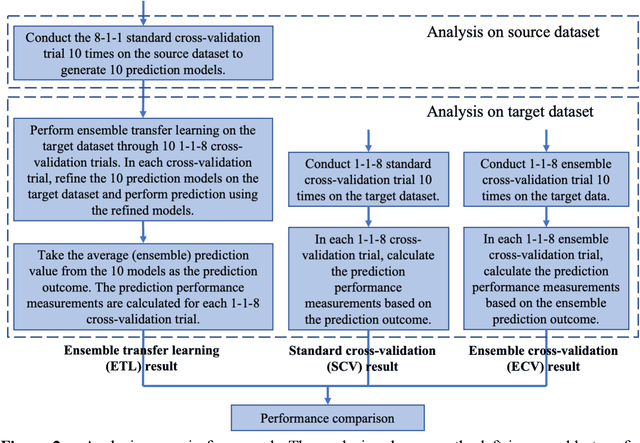
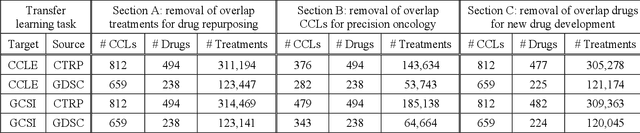
Transfer learning has been shown to be effective in many applications in which training data for the target problem are limited but data for a related (source) problem are abundant. In this paper, we apply transfer learning to the prediction of anti-cancer drug response. Previous transfer learning studies for drug response prediction focused on building models that predict the response of tumor cells to a specific drug treatment. We target the more challenging task of building general prediction models that can make predictions for both new tumor cells and new drugs. We apply the classic transfer learning framework that trains a prediction model on the source dataset and refines it on the target dataset, and extends the framework through ensemble. The ensemble transfer learning pipeline is implemented using LightGBM and two deep neural network (DNN) models with different architectures. Uniquely, we investigate its power for three application settings including drug repurposing, precision oncology, and new drug development, through different data partition schemes in cross-validation. We test the proposed ensemble transfer learning on benchmark in vitro drug screening datasets, taking one dataset as the source domain and another dataset as the target domain. The analysis results demonstrate the benefit of applying ensemble transfer learning for predicting anti-cancer drug response in all three applications with both LightGBM and DNN models. Compared between the different prediction models, a DNN model with two subnetworks for the inputs of tumor features and drug features separately outperforms LightGBM and the other DNN model that concatenates tumor features and drug features for input in the drug repurposing and precision oncology applications. In the more challenging application of new drug development, LightGBM performs better than the other two DNN models.
A Systematic Approach to Featurization for Cancer Drug Sensitivity Predictions with Deep Learning
May 04, 2020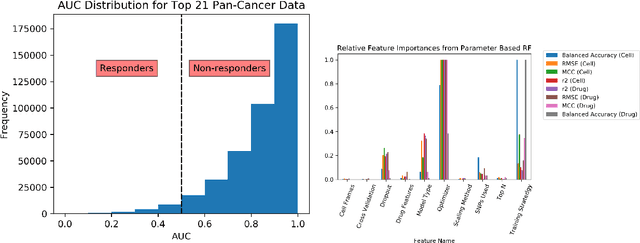
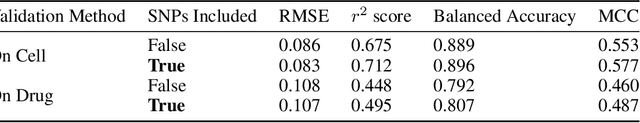

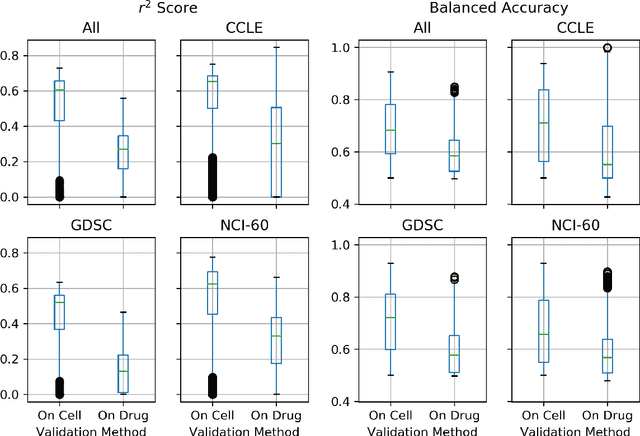
By combining various cancer cell line (CCL) drug screening panels, the size of the data has grown significantly to begin understanding how advances in deep learning can advance drug response predictions. In this paper we train >35,000 neural network models, sweeping over common featurization techniques. We found the RNA-seq to be highly redundant and informative even with subsets larger than 128 features. We found the inclusion of single nucleotide polymorphisms (SNPs) coded as count matrices improved model performance significantly, and no substantial difference in model performance with respect to molecular featurization between the common open source MOrdred descriptors and Dragon7 descriptors. Alongside this analysis, we outline data integration between CCL screening datasets and present evidence that new metrics and imbalanced data techniques, as well as advances in data standardization, need to be developed.