 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeScalable Runtime Architecture for Data-driven, Hybrid HPC and ML Workflow Applications
Mar 17, 2025Hybrid workflows combining traditional HPC and novel ML methodologies are transforming scientific computing. This paper presents the architecture and implementation of a scalable runtime system that extends RADICAL-Pilot with service-based execution to support AI-out-HPC workflows. Our runtime system enables distributed ML capabilities, efficient resource management, and seamless HPC/ML coupling across local and remote platforms. Preliminary experimental results show that our approach manages concurrent execution of ML models across local and remote HPC/cloud resources with minimal architectural overheads. This lays the foundation for prototyping three representative data-driven workflow applications and executing them at scale on leadership-class HPC platforms.
Protein-Ligand Docking Surrogate Models: A SARS-CoV-2 Benchmark for Deep Learning Accelerated Virtual Screening
Jun 30, 2021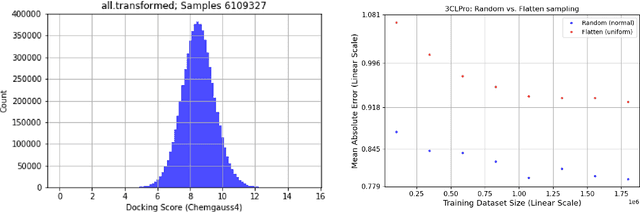

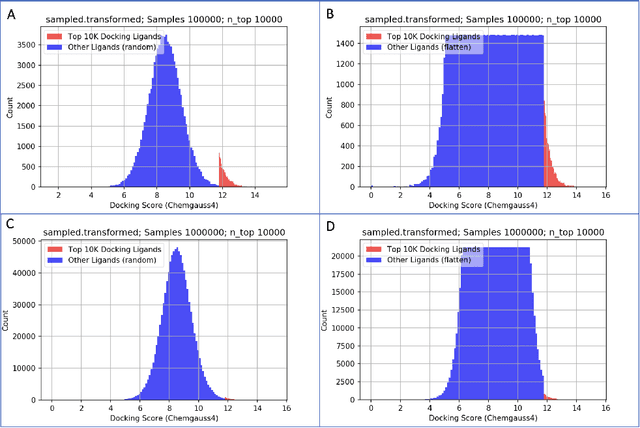
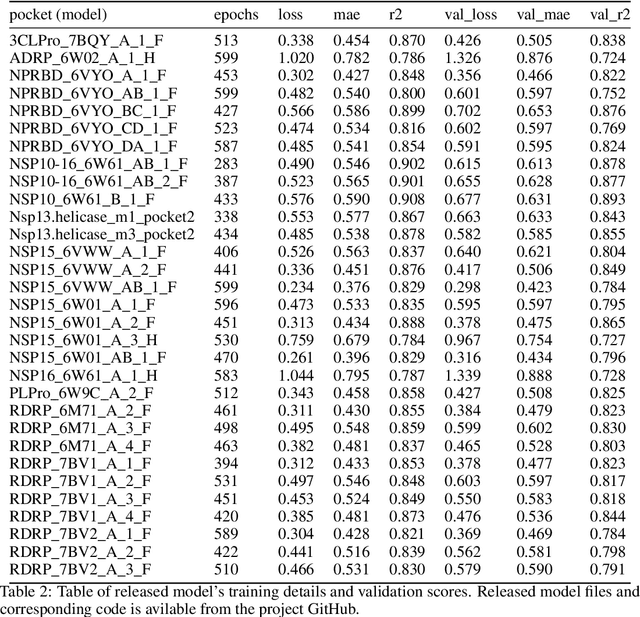
We propose a benchmark to study surrogate model accuracy for protein-ligand docking. We share a dataset consisting of 200 million 3D complex structures and 2D structure scores across a consistent set of 13 million "in-stock" molecules over 15 receptors, or binding sites, across the SARS-CoV-2 proteome. Our work shows surrogate docking models have six orders of magnitude more throughput than standard docking protocols on the same supercomputer node types. We demonstrate the power of high-speed surrogate models by running each target against 1 billion molecules in under a day (50k predictions per GPU seconds). We showcase a workflow for docking utilizing surrogate ML models as a pre-filter. Our workflow is ten times faster at screening a library of compounds than the standard technique, with an error rate less than 0.01\% of detecting the underlying best scoring 0.1\% of compounds. Our analysis of the speedup explains that to screen more molecules under a docking paradigm, another order of magnitude speedup must come from model accuracy rather than computing speed (which, if increased, will not anymore alter our throughput to screen molecules). We believe this is strong evidence for the community to begin focusing on improving the accuracy of surrogate models to improve the ability to screen massive compound libraries 100x or even 1000x faster than current techniques.
Pandemic Drugs at Pandemic Speed: Accelerating COVID-19 Drug Discovery with Hybrid Machine Learning- and Physics-based Simulations on High Performance Computers
Mar 04, 2021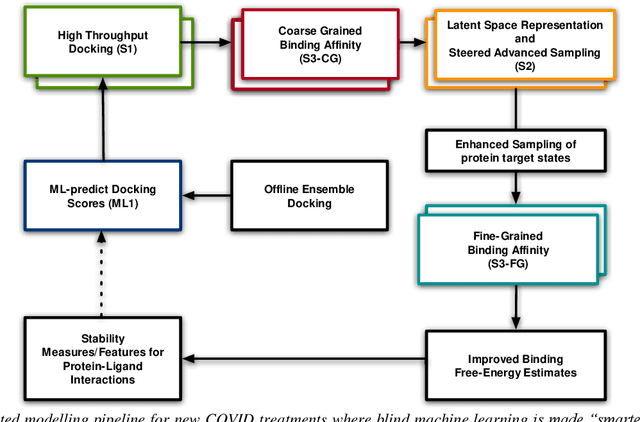

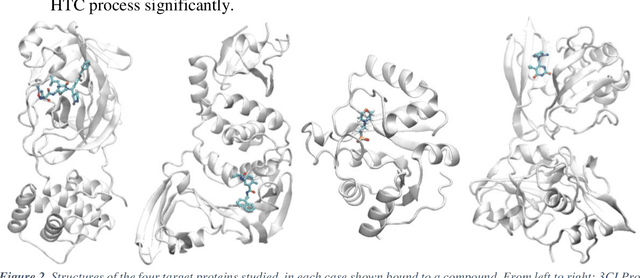
The race to meet the challenges of the global pandemic has served as a reminder that the existing drug discovery process is expensive, inefficient and slow. There is a major bottleneck screening the vast number of potential small molecules to shortlist lead compounds for antiviral drug development. New opportunities to accelerate drug discovery lie at the interface between machine learning methods, in this case developed for linear accelerators, and physics-based methods. The two in silico methods, each have their own advantages and limitations which, interestingly, complement each other. Here, we present an innovative method that combines both approaches to accelerate drug discovery. The scale of the resulting workflow is such that it is dependent on high performance computing. We have demonstrated the applicability of this workflow on four COVID-19 target proteins and our ability to perform the required large-scale calculations to identify lead compounds on a variety of supercomputers.