 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking community drug response prediction models: datasets, models, tools, and metrics for cross-dataset generalization analysis
Mar 18, 2025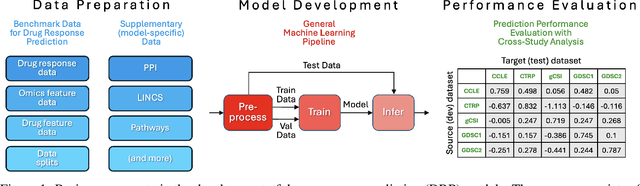
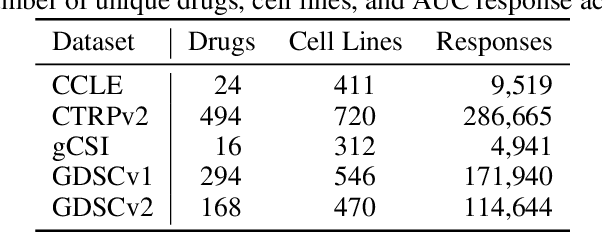
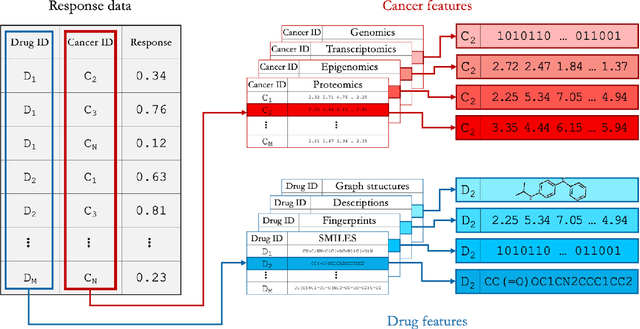

Deep learning (DL) and machine learning (ML) models have shown promise in drug response prediction (DRP), yet their ability to generalize across datasets remains an open question, raising concerns about their real-world applicability. Due to the lack of standardized benchmarking approaches, model evaluations and comparisons often rely on inconsistent datasets and evaluation criteria, making it difficult to assess true predictive capabilities. In this work, we introduce a benchmarking framework for evaluating cross-dataset prediction generalization in DRP models. Our framework incorporates five publicly available drug screening datasets, six standardized DRP models, and a scalable workflow for systematic evaluation. To assess model generalization, we introduce a set of evaluation metrics that quantify both absolute performance (e.g., predictive accuracy across datasets) and relative performance (e.g., performance drop compared to within-dataset results), enabling a more comprehensive assessment of model transferability. Our results reveal substantial performance drops when models are tested on unseen datasets, underscoring the importance of rigorous generalization assessments. While several models demonstrate relatively strong cross-dataset generalization, no single model consistently outperforms across all datasets. Furthermore, we identify CTRPv2 as the most effective source dataset for training, yielding higher generalization scores across target datasets. By sharing this standardized evaluation framework with the community, our study aims to establish a rigorous foundation for model comparison, and accelerate the development of robust DRP models for real-world applications.
Variational and Explanatory Neural Networks for Encoding Cancer Profiles and Predicting Drug Responses
Jul 05, 2024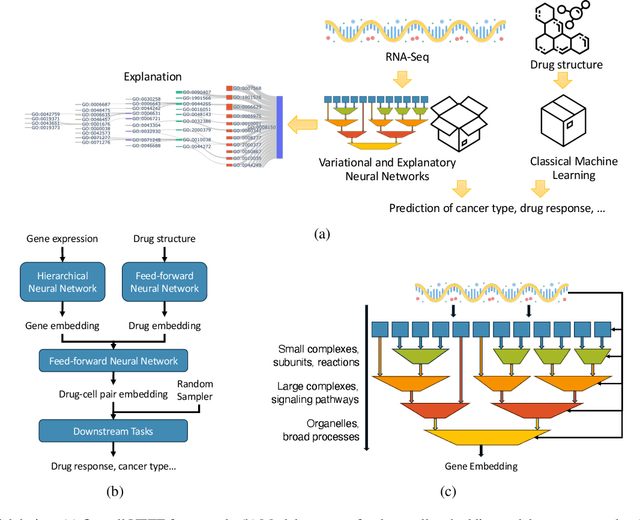

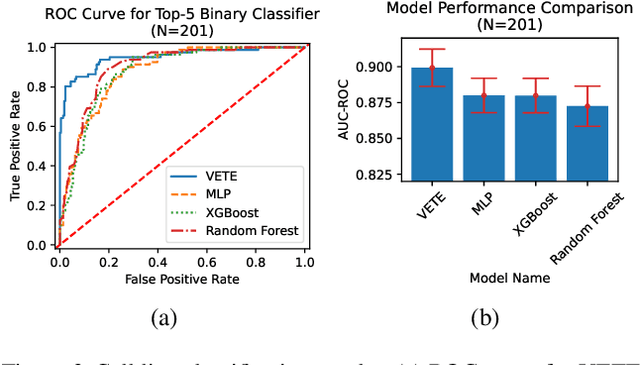
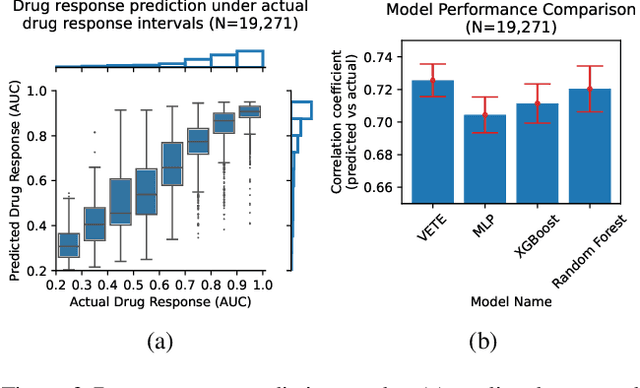
Human cancers present a significant public health challenge and require the discovery of novel drugs through translational research. Transcriptomics profiling data that describes molecular activities in tumors and cancer cell lines are widely utilized for predicting anti-cancer drug responses. However, existing AI models face challenges due to noise in transcriptomics data and lack of biological interpretability. To overcome these limitations, we introduce VETE (Variational and Explanatory Transcriptomics Encoder), a novel neural network framework that incorporates a variational component to mitigate noise effects and integrates traceable gene ontology into the neural network architecture for encoding cancer transcriptomics data. Key innovations include a local interpretability-guided method for identifying ontology paths, a visualization tool to elucidate biological mechanisms of drug responses, and the application of centralized large scale hyperparameter optimization. VETE demonstrated robust accuracy in cancer cell line classification and drug response prediction. Additionally, it provided traceable biological explanations for both tasks and offers insights into the mechanisms underlying its predictions. VETE bridges the gap between AI-driven predictions and biologically meaningful insights in cancer research, which represents a promising advancement in the field.
Deep learning methods for drug response prediction in cancer: predominant and emerging trends
Nov 18, 2022Cancer claims millions of lives yearly worldwide. While many therapies have been made available in recent years, by in large cancer remains unsolved. Exploiting computational predictive models to study and treat cancer holds great promise in improving drug development and personalized design of treatment plans, ultimately suppressing tumors, alleviating suffering, and prolonging lives of patients. A wave of recent papers demonstrates promising results in predicting cancer response to drug treatments while utilizing deep learning methods. These papers investigate diverse data representations, neural network architectures, learning methodologies, and evaluations schemes. However, deciphering promising predominant and emerging trends is difficult due to the variety of explored methods and lack of standardized framework for comparing drug response prediction models. To obtain a comprehensive landscape of deep learning methods, we conducted an extensive search and analysis of deep learning models that predict the response to single drug treatments. A total of 60 deep learning-based models have been curated and summary plots were generated. Based on the analysis, observable patterns and prevalence of methods have been revealed. This review allows to better understand the current state of the field and identify major challenges and promising solution paths.
Protein-Ligand Docking Surrogate Models: A SARS-CoV-2 Benchmark for Deep Learning Accelerated Virtual Screening
Jun 30, 2021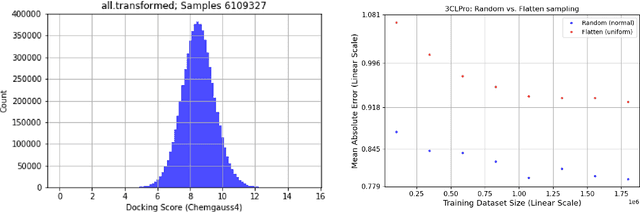

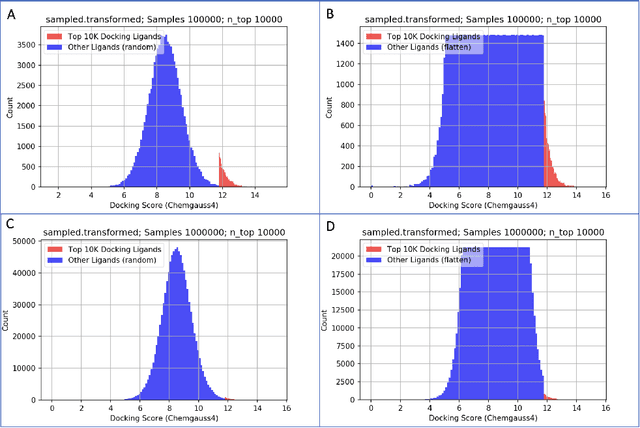
We propose a benchmark to study surrogate model accuracy for protein-ligand docking. We share a dataset consisting of 200 million 3D complex structures and 2D structure scores across a consistent set of 13 million "in-stock" molecules over 15 receptors, or binding sites, across the SARS-CoV-2 proteome. Our work shows surrogate docking models have six orders of magnitude more throughput than standard docking protocols on the same supercomputer node types. We demonstrate the power of high-speed surrogate models by running each target against 1 billion molecules in under a day (50k predictions per GPU seconds). We showcase a workflow for docking utilizing surrogate ML models as a pre-filter. Our workflow is ten times faster at screening a library of compounds than the standard technique, with an error rate less than 0.01\% of detecting the underlying best scoring 0.1\% of compounds. Our analysis of the speedup explains that to screen more molecules under a docking paradigm, another order of magnitude speedup must come from model accuracy rather than computing speed (which, if increased, will not anymore alter our throughput to screen molecules). We believe this is strong evidence for the community to begin focusing on improving the accuracy of surrogate models to improve the ability to screen massive compound libraries 100x or even 1000x faster than current techniques.
Pandemic Drugs at Pandemic Speed: Accelerating COVID-19 Drug Discovery with Hybrid Machine Learning- and Physics-based Simulations on High Performance Computers
Mar 04, 2021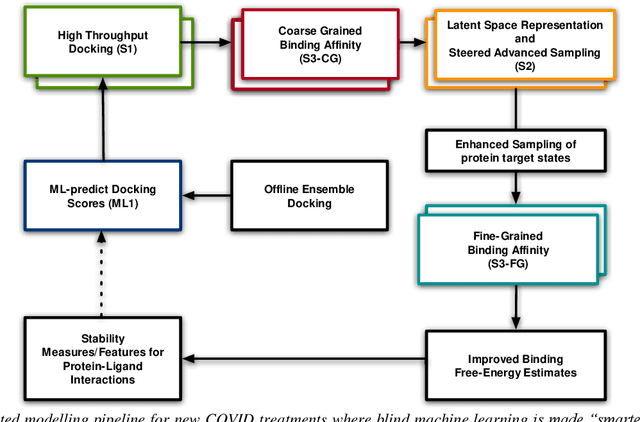

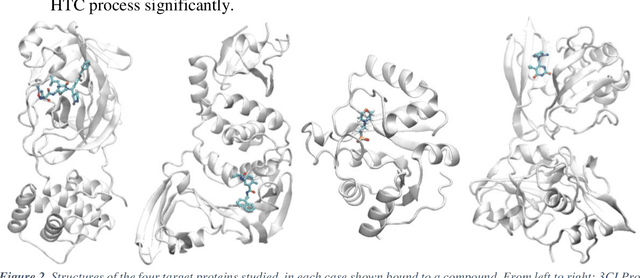
The race to meet the challenges of the global pandemic has served as a reminder that the existing drug discovery process is expensive, inefficient and slow. There is a major bottleneck screening the vast number of potential small molecules to shortlist lead compounds for antiviral drug development. New opportunities to accelerate drug discovery lie at the interface between machine learning methods, in this case developed for linear accelerators, and physics-based methods. The two in silico methods, each have their own advantages and limitations which, interestingly, complement each other. Here, we present an innovative method that combines both approaches to accelerate drug discovery. The scale of the resulting workflow is such that it is dependent on high performance computing. We have demonstrated the applicability of this workflow on four COVID-19 target proteins and our ability to perform the required large-scale calculations to identify lead compounds on a variety of supercomputers.
Learning Curves for Drug Response Prediction in Cancer Cell Lines
Nov 25, 2020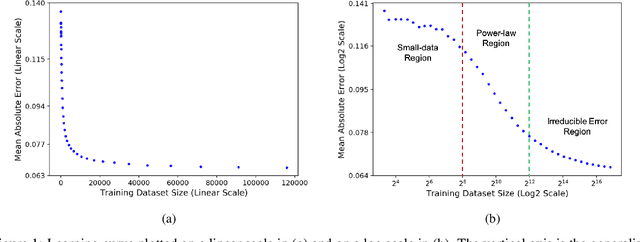
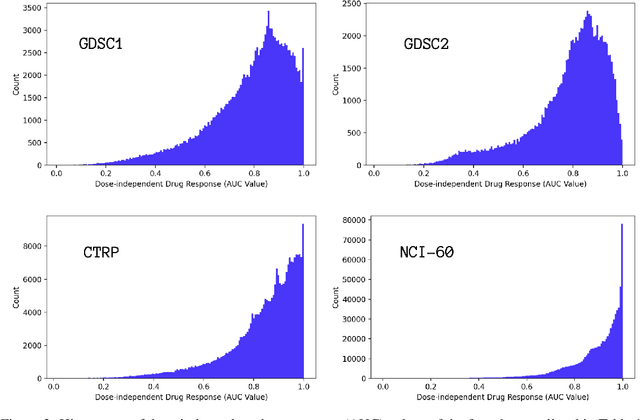
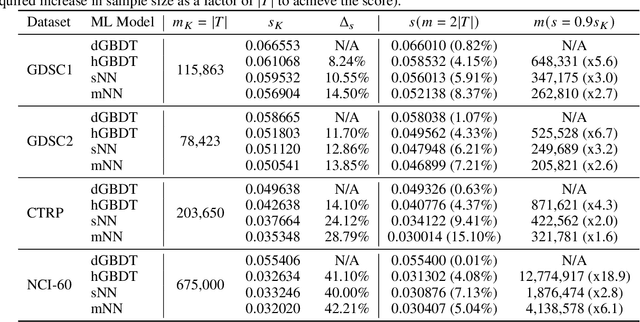
Motivated by the size of cell line drug sensitivity data, researchers have been developing machine learning (ML) models for predicting drug response to advance cancer treatment. As drug sensitivity studies continue generating data, a common question is whether the proposed predictors can further improve the generalization performance with more training data. We utilize empirical learning curves for evaluating and comparing the data scaling properties of two neural networks (NNs) and two gradient boosting decision tree (GBDT) models trained on four drug screening datasets. The learning curves are accurately fitted to a power law model, providing a framework for assessing the data scaling behavior of these predictors. The curves demonstrate that no single model dominates in terms of prediction performance across all datasets and training sizes, suggesting that the shape of these curves depends on the unique model-dataset pair. The multi-input NN (mNN), in which gene expressions and molecular drug descriptors are input into separate subnetworks, outperforms a single-input NN (sNN), where the cell and drug features are concatenated for the input layer. In contrast, a GBDT with hyperparameter tuning exhibits superior performance as compared with both NNs at the lower range of training sizes for two of the datasets, whereas the mNN performs better at the higher range of training sizes. Moreover, the trajectory of the curves suggests that increasing the sample size is expected to further improve prediction scores of both NNs. These observations demonstrate the benefit of using learning curves to evaluate predictors, providing a broader perspective on the overall data scaling characteristics. The fitted power law curves provide a forward-looking performance metric and can serve as a co-design tool to guide experimental biologists and computational scientists in the design of future experiments.
Ensemble Transfer Learning for the Prediction of Anti-Cancer Drug Response
May 13, 2020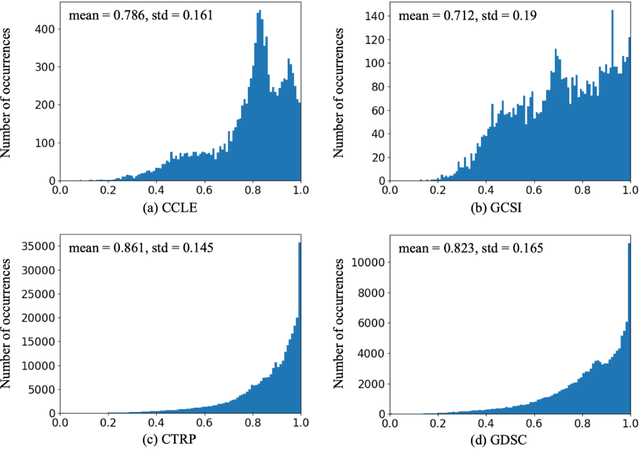
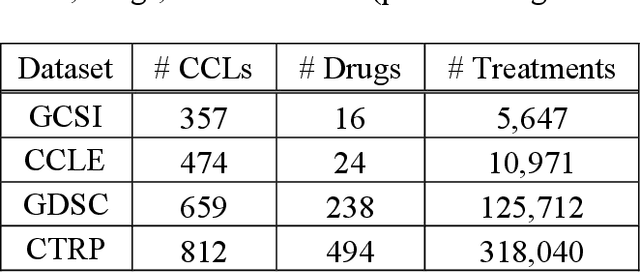
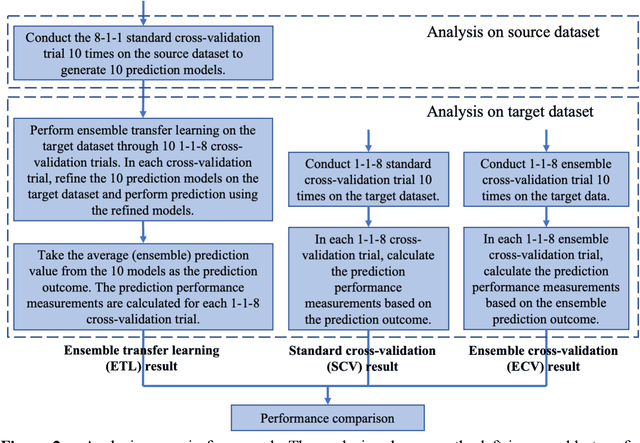
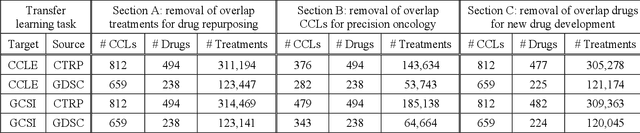
Transfer learning has been shown to be effective in many applications in which training data for the target problem are limited but data for a related (source) problem are abundant. In this paper, we apply transfer learning to the prediction of anti-cancer drug response. Previous transfer learning studies for drug response prediction focused on building models that predict the response of tumor cells to a specific drug treatment. We target the more challenging task of building general prediction models that can make predictions for both new tumor cells and new drugs. We apply the classic transfer learning framework that trains a prediction model on the source dataset and refines it on the target dataset, and extends the framework through ensemble. The ensemble transfer learning pipeline is implemented using LightGBM and two deep neural network (DNN) models with different architectures. Uniquely, we investigate its power for three application settings including drug repurposing, precision oncology, and new drug development, through different data partition schemes in cross-validation. We test the proposed ensemble transfer learning on benchmark in vitro drug screening datasets, taking one dataset as the source domain and another dataset as the target domain. The analysis results demonstrate the benefit of applying ensemble transfer learning for predicting anti-cancer drug response in all three applications with both LightGBM and DNN models. Compared between the different prediction models, a DNN model with two subnetworks for the inputs of tumor features and drug features separately outperforms LightGBM and the other DNN model that concatenates tumor features and drug features for input in the drug repurposing and precision oncology applications. In the more challenging application of new drug development, LightGBM performs better than the other two DNN models.
A Systematic Approach to Featurization for Cancer Drug Sensitivity Predictions with Deep Learning
May 04, 2020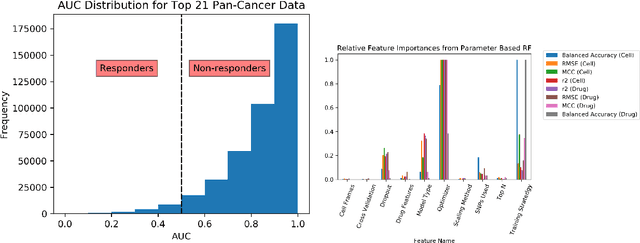
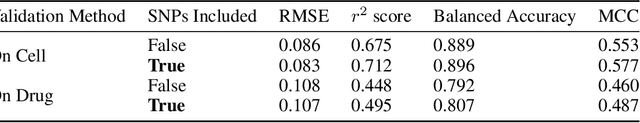

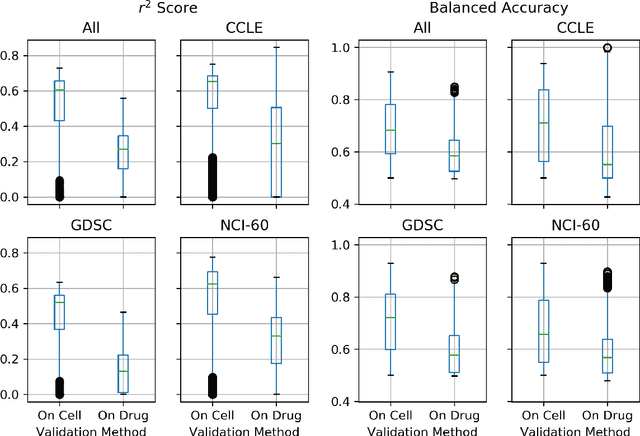
By combining various cancer cell line (CCL) drug screening panels, the size of the data has grown significantly to begin understanding how advances in deep learning can advance drug response predictions. In this paper we train >35,000 neural network models, sweeping over common featurization techniques. We found the RNA-seq to be highly redundant and informative even with subsets larger than 128 features. We found the inclusion of single nucleotide polymorphisms (SNPs) coded as count matrices improved model performance significantly, and no substantial difference in model performance with respect to molecular featurization between the common open source MOrdred descriptors and Dragon7 descriptors. Alongside this analysis, we outline data integration between CCL screening datasets and present evidence that new metrics and imbalanced data techniques, as well as advances in data standardization, need to be developed.