 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRethinking ANN-based Retrieval: Multifaceted Learnable Index for Large-scale Recommendation System
Feb 18, 2026Approximate nearest neighbor (ANN) search is widely used in the retrieval stage of large-scale recommendation systems. In this stage, candidate items are indexed using their learned embedding vectors, and ANN search is executed for each user (or item) query to retrieve a set of relevant items. However, ANN-based retrieval has two key limitations. First, item embeddings and their indices are typically learned in separate stages: indexing is often performed offline after embeddings are trained, which can yield suboptimal retrieval quality-especially for newly created items. Second, although ANN offers sublinear query time, it must still be run for every request, incurring substantial computation cost at industry scale. In this paper, we propose MultiFaceted Learnable Index (MFLI), a scalable, real-time retrieval paradigm that learns multifaceted item embeddings and indices within a unified framework and eliminates ANN search at serving time. Specifically, we construct a multifaceted hierarchical codebook via residual quantization of item embeddings and co-train the codebook with the embeddings. We further introduce an efficient multifaceted indexing structure and mechanisms that support real-time updates. At serving time, the learned hierarchical indices are used directly to identify relevant items, avoiding ANN search altogether. Extensive experiments on real-world data with billions of users show that MFLI improves recall on engagement tasks by up to 11.8\%, cold-content delivery by up to 57.29\%, and semantic relevance by 13.5\% compared with prior state-of-the-art methods. We also deploy MFLI in the system and report online experimental results demonstrating improved engagement, less popularity bias, and higher serving efficiency.
Monte Carlo Tree Diffusion with Multiple Experts for Protein Design
Sep 19, 2025The goal of protein design is to generate amino acid sequences that fold into functional structures with desired properties. Prior methods combining autoregressive language models with Monte Carlo Tree Search (MCTS) struggle with long-range dependencies and suffer from an impractically large search space. We propose MCTD-ME, Monte Carlo Tree Diffusion with Multiple Experts, which integrates masked diffusion models with tree search to enable multi-token planning and efficient exploration. Unlike autoregressive planners, MCTD-ME uses biophysical-fidelity-enhanced diffusion denoising as the rollout engine, jointly revising multiple positions and scaling to large sequence spaces. It further leverages experts of varying capacities to enrich exploration, guided by a pLDDT-based masking schedule that targets low-confidence regions while preserving reliable residues. We propose a novel multi-expert selection rule (PH-UCT-ME) extends predictive-entropy UCT to expert ensembles. On the inverse folding task (CAMEO and PDB benchmarks), MCTD-ME outperforms single-expert and unguided baselines in both sequence recovery (AAR) and structural similarity (scTM), with gains increasing for longer proteins and benefiting from multi-expert guidance. More generally, the framework is model-agnostic and applicable beyond inverse folding, including de novo protein engineering and multi-objective molecular generation.
Bidirectional Hierarchical Protein Multi-Modal Representation Learning
Apr 07, 2025Protein representation learning is critical for numerous biological tasks. Recently, large transformer-based protein language models (pLMs) pretrained on large scale protein sequences have demonstrated significant success in sequence-based tasks. However, pLMs lack structural information. Conversely, graph neural networks (GNNs) designed to leverage 3D structural information have shown promising generalization in protein-related prediction tasks, but their effectiveness is often constrained by the scarcity of labeled structural data. Recognizing that sequence and structural representations are complementary perspectives of the same protein entity, we propose a multimodal bidirectional hierarchical fusion framework to effectively merge these modalities. Our framework employs attention and gating mechanisms to enable effective interaction between pLMs-generated sequential representations and GNN-extracted structural features, improving information exchange and enhancement across layers of the neural network. Based on the framework, we further introduce local Bi-Hierarchical Fusion with gating and global Bi-Hierarchical Fusion with multihead self-attention approaches. Through extensive experiments on a diverse set of protein-related tasks, our method demonstrates consistent improvements over strong baselines and existing fusion techniques in a variety of protein representation learning benchmarks, including react (enzyme/EC classification), model quality assessment (MQA), protein-ligand binding affinity prediction (LBA), protein-protein binding site prediction (PPBS), and B cell epitopes prediction (BCEs). Our method establishes a new state-of-the-art for multimodal protein representation learning, emphasizing the efficacy of BIHIERARCHICAL FUSION in bridging sequence and structural modalities.
DrugImproverGPT: A Large Language Model for Drug Optimization with Fine-Tuning via Structured Policy Optimization
Feb 11, 2025Finetuning a Large Language Model (LLM) is crucial for generating results towards specific objectives. This research delves into the realm of drug optimization and introduce a novel reinforcement learning algorithm to finetune a drug optimization LLM-based generative model, enhancing the original drug across target objectives, while retains the beneficial chemical properties of the original drug. This work is comprised of two primary components: (1) DrugImprover: A framework tailored for improving robustness and efficiency in drug optimization. It includes a LLM designed for drug optimization and a novel Structured Policy Optimization (SPO) algorithm, which is theoretically grounded. This algorithm offers a unique perspective for fine-tuning the LLM-based generative model by aligning the improvement of the generated molecule with the input molecule under desired objectives. (2) A dataset of 1 million compounds, each with OEDOCK docking scores on 5 human proteins associated with cancer cells and 24 binding sites from SARS-CoV-2 virus. We conduct a comprehensive evaluation of SPO and demonstrate its effectiveness in improving the original drug across target properties. Our code and dataset will be publicly available at: https://github.com/xuefeng-cs/DrugImproverGPT.
ScaffoldGPT: A Scaffold-based Large Language Model for Drug Improvement
Feb 09, 2025Drug optimization has become increasingly crucial in light of fast-mutating virus strains and drug-resistant cancer cells. Nevertheless, it remains challenging as it necessitates retaining the beneficial properties of the original drug while simultaneously enhancing desired attributes beyond its scope. In this work, we aim to tackle this challenge by introducing ScaffoldGPT, a novel Large Language Model (LLM) designed for drug optimization based on molecular scaffolds. Our work comprises three key components: (1) A three-stage drug optimization approach that integrates pretraining, finetuning, and decoding optimization. (2) A uniquely designed two-phase incremental training approach for pre-training the drug optimization LLM-based generator on molecule scaffold with enhanced performance. (3) A token-level decoding optimization strategy, TOP-N, that enabling controlled, reward-guided generation using pretrained/finetuned LLMs. Finally, by conducting a comprehensive evaluation on COVID and cancer benchmarks, we demonstrate that SCAFFOLDGPT outperforms the competing baselines in drug optimization benchmarks, while excelling in preserving the original functional scaffold and enhancing desired properties.
Entropy-Reinforced Planning with Large Language Models for Drug Discovery
Jun 11, 2024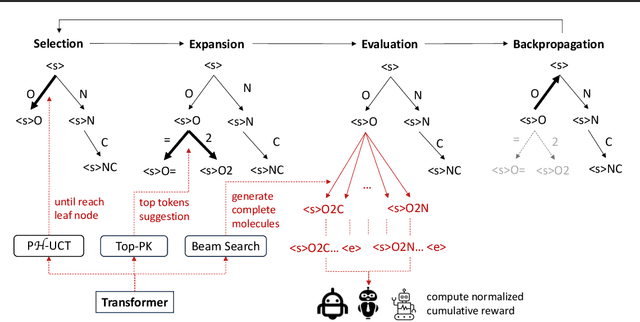
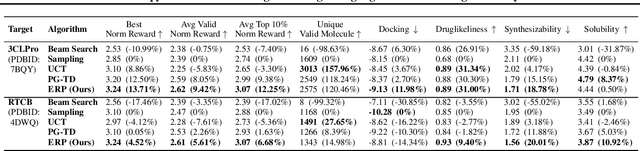
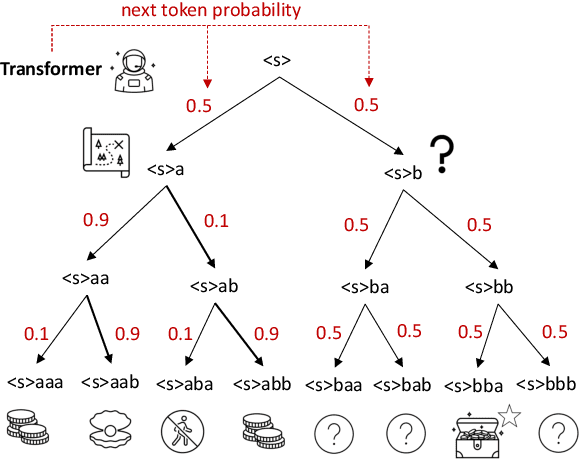
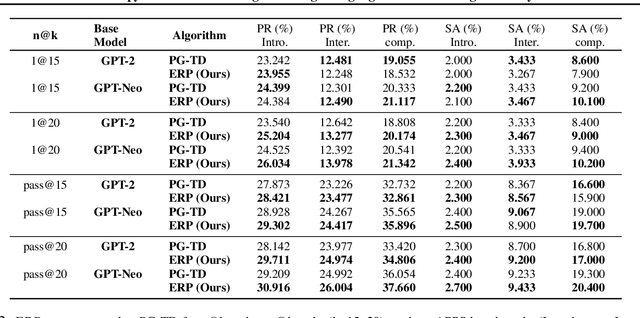
The objective of drug discovery is to identify chemical compounds that possess specific pharmaceutical properties toward a binding target. Existing large language models (LLMS) can achieve high token matching scores in terms of likelihood for molecule generation. However, relying solely on LLM decoding often results in the generation of molecules that are either invalid due to a single misused token, or suboptimal due to unbalanced exploration and exploitation as a consequence of the LLMs prior experience. Here we propose ERP, Entropy-Reinforced Planning for Transformer Decoding, which employs an entropy-reinforced planning algorithm to enhance the Transformer decoding process and strike a balance between exploitation and exploration. ERP aims to achieve improvements in multiple properties compared to direct sampling from the Transformer. We evaluated ERP on the SARS-CoV-2 virus (3CLPro) and human cancer cell target protein (RTCB) benchmarks and demonstrated that, in both benchmarks, ERP consistently outperforms the current state-of-the-art algorithm by 1-5 percent, and baselines by 5-10 percent, respectively. Moreover, such improvement is robust across Transformer models trained with different objectives. Finally, to further illustrate the capabilities of ERP, we tested our algorithm on three code generation benchmarks and outperformed the current state-of-the-art approach as well. Our code is publicly available at: https://github.com/xuefeng-cs/ERP.
Feature Weaken: Vicinal Data Augmentation for Classification
Nov 20, 2022Deep learning usually relies on training large-scale data samples to achieve better performance. However, over-fitting based on training data always remains a problem. Scholars have proposed various strategies, such as feature dropping and feature mixing, to improve the generalization continuously. For the same purpose, we subversively propose a novel training method, Feature Weaken, which can be regarded as a data augmentation method. Feature Weaken constructs the vicinal data distribution with the same cosine similarity for model training by weakening features of the original samples. In especially, Feature Weaken changes the spatial distribution of samples, adjusts sample boundaries, and reduces the gradient optimization value of back-propagation. This work can not only improve the classification performance and generalization of the model, but also stabilize the model training and accelerate the model convergence. We conduct extensive experiments on classical deep convolution neural models with five common image classification datasets and the Bert model with four common text classification datasets. Compared with the classical models or the generalization improvement methods, such as Dropout, Mixup, Cutout, and CutMix, Feature Weaken shows good compatibility and performance. We also use adversarial samples to perform the robustness experiments, and the results show that Feature Weaken is effective in improving the robustness of the model.
Learning Curves for Drug Response Prediction in Cancer Cell Lines
Nov 25, 2020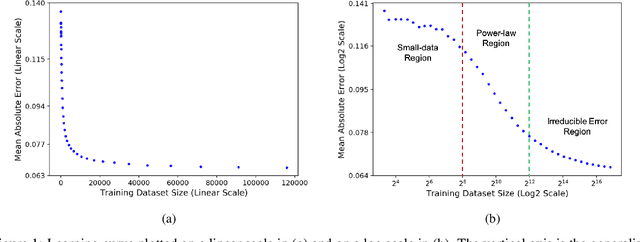
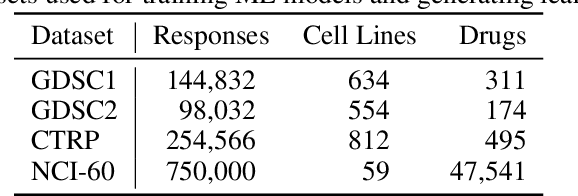
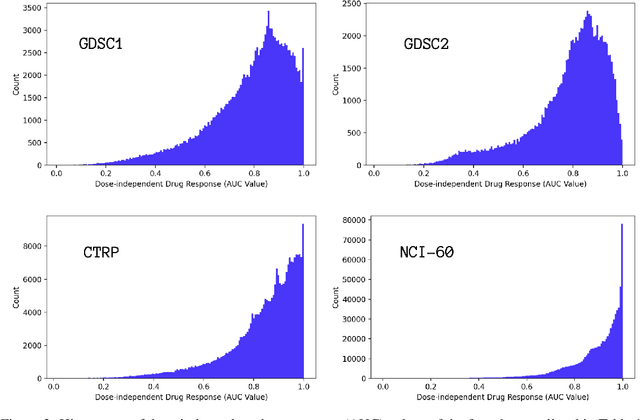
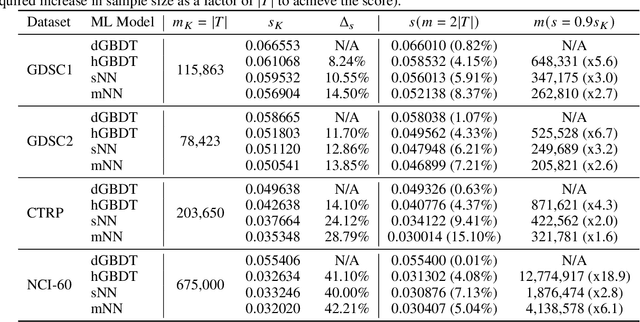
Motivated by the size of cell line drug sensitivity data, researchers have been developing machine learning (ML) models for predicting drug response to advance cancer treatment. As drug sensitivity studies continue generating data, a common question is whether the proposed predictors can further improve the generalization performance with more training data. We utilize empirical learning curves for evaluating and comparing the data scaling properties of two neural networks (NNs) and two gradient boosting decision tree (GBDT) models trained on four drug screening datasets. The learning curves are accurately fitted to a power law model, providing a framework for assessing the data scaling behavior of these predictors. The curves demonstrate that no single model dominates in terms of prediction performance across all datasets and training sizes, suggesting that the shape of these curves depends on the unique model-dataset pair. The multi-input NN (mNN), in which gene expressions and molecular drug descriptors are input into separate subnetworks, outperforms a single-input NN (sNN), where the cell and drug features are concatenated for the input layer. In contrast, a GBDT with hyperparameter tuning exhibits superior performance as compared with both NNs at the lower range of training sizes for two of the datasets, whereas the mNN performs better at the higher range of training sizes. Moreover, the trajectory of the curves suggests that increasing the sample size is expected to further improve prediction scores of both NNs. These observations demonstrate the benefit of using learning curves to evaluate predictors, providing a broader perspective on the overall data scaling characteristics. The fitted power law curves provide a forward-looking performance metric and can serve as a co-design tool to guide experimental biologists and computational scientists in the design of future experiments.