 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSurface-based Molecular Design with Multi-modal Flow Matching
Jan 08, 2026Therapeutic peptides show promise in targeting previously undruggable binding sites, with recent advancements in deep generative models enabling full-atom peptide co-design for specific protein receptors. However, the critical role of molecular surfaces in protein-protein interactions (PPIs) has been underexplored. To bridge this gap, we propose an omni-design peptides generation paradigm, called SurfFlow, a novel surface-based generative algorithm that enables comprehensive co-design of sequence, structure, and surface for peptides. SurfFlow employs a multi-modality conditional flow matching (CFM) architecture to learn distributions of surface geometries and biochemical properties, enhancing peptide binding accuracy. Evaluated on the comprehensive PepMerge benchmark, SurfFlow consistently outperforms full-atom baselines across all metrics. These results highlight the advantages of considering molecular surfaces in de novo peptide discovery and demonstrate the potential of integrating multiple protein modalities for more effective therapeutic peptide discovery.
From Supervision to Exploration: What Does Protein Language Model Learn During Reinforcement Learning?
Oct 02, 2025Protein language models (PLMs) have advanced computational protein science through large-scale pretraining and scalable architectures. In parallel, reinforcement learning (RL) has broadened exploration and enabled precise multi-objective optimization in protein design. Yet whether RL can push PLMs beyond their pretraining priors to uncover latent sequence-structure-function rules remains unclear. We address this by pairing RL with PLMs across four domains: antimicrobial peptide design, kinase variant optimization, antibody engineering, and inverse folding. Using diverse RL algorithms and model classes, we ask if RL improves sampling efficiency and, more importantly, if it reveals capabilities not captured by supervised learning. Across benchmarks, RL consistently boosts success rates and sample efficiency. Performance follows a three-factor interaction: task headroom, reward fidelity, and policy capacity jointly determine gains. When rewards are accurate and informative, policies have sufficient capacity, and tasks leave room beyond supervised baselines, improvements scale; when rewards are noisy or capacity is constrained, gains saturate despite exploration. This view yields practical guidance for RL in protein design: prioritize reward modeling and calibration before scaling policy size, match algorithm and regularization strength to task difficulty, and allocate capacity where marginal gains are largest. Implementation is available at https://github.com/chq1155/RL-PLM.
Monte Carlo Tree Diffusion with Multiple Experts for Protein Design
Sep 19, 2025The goal of protein design is to generate amino acid sequences that fold into functional structures with desired properties. Prior methods combining autoregressive language models with Monte Carlo Tree Search (MCTS) struggle with long-range dependencies and suffer from an impractically large search space. We propose MCTD-ME, Monte Carlo Tree Diffusion with Multiple Experts, which integrates masked diffusion models with tree search to enable multi-token planning and efficient exploration. Unlike autoregressive planners, MCTD-ME uses biophysical-fidelity-enhanced diffusion denoising as the rollout engine, jointly revising multiple positions and scaling to large sequence spaces. It further leverages experts of varying capacities to enrich exploration, guided by a pLDDT-based masking schedule that targets low-confidence regions while preserving reliable residues. We propose a novel multi-expert selection rule (PH-UCT-ME) extends predictive-entropy UCT to expert ensembles. On the inverse folding task (CAMEO and PDB benchmarks), MCTD-ME outperforms single-expert and unguided baselines in both sequence recovery (AAR) and structural similarity (scTM), with gains increasing for longer proteins and benefiting from multi-expert guidance. More generally, the framework is model-agnostic and applicable beyond inverse folding, including de novo protein engineering and multi-objective molecular generation.
Bidirectional Hierarchical Protein Multi-Modal Representation Learning
Apr 07, 2025Protein representation learning is critical for numerous biological tasks. Recently, large transformer-based protein language models (pLMs) pretrained on large scale protein sequences have demonstrated significant success in sequence-based tasks. However, pLMs lack structural information. Conversely, graph neural networks (GNNs) designed to leverage 3D structural information have shown promising generalization in protein-related prediction tasks, but their effectiveness is often constrained by the scarcity of labeled structural data. Recognizing that sequence and structural representations are complementary perspectives of the same protein entity, we propose a multimodal bidirectional hierarchical fusion framework to effectively merge these modalities. Our framework employs attention and gating mechanisms to enable effective interaction between pLMs-generated sequential representations and GNN-extracted structural features, improving information exchange and enhancement across layers of the neural network. Based on the framework, we further introduce local Bi-Hierarchical Fusion with gating and global Bi-Hierarchical Fusion with multihead self-attention approaches. Through extensive experiments on a diverse set of protein-related tasks, our method demonstrates consistent improvements over strong baselines and existing fusion techniques in a variety of protein representation learning benchmarks, including react (enzyme/EC classification), model quality assessment (MQA), protein-ligand binding affinity prediction (LBA), protein-protein binding site prediction (PPBS), and B cell epitopes prediction (BCEs). Our method establishes a new state-of-the-art for multimodal protein representation learning, emphasizing the efficacy of BIHIERARCHICAL FUSION in bridging sequence and structural modalities.
Leveraging Multimodal Protein Representations to Predict Protein Melting Temperatures
Dec 05, 2024



Accurately predicting protein melting temperature changes (Delta Tm) is fundamental for assessing protein stability and guiding protein engineering. Leveraging multi-modal protein representations has shown great promise in capturing the complex relationships among protein sequences, structures, and functions. In this study, we develop models based on powerful protein language models, including ESM-2, ESM-3, SaProt, and AlphaFold, using various feature extraction methods to enhance prediction accuracy. By utilizing the ESM-3 model, we achieve a new state-of-the-art performance on the s571 test dataset, obtaining a Pearson correlation coefficient (PCC) of 0.50. Furthermore, we conduct a fair evaluation to compare the performance of different protein language models in the Delta Tm prediction task. Our results demonstrate that integrating multi-modal protein representations could advance the prediction of protein melting temperatures.
InstructBio: A Large-scale Semi-supervised Learning Paradigm for Biochemical Problems
Apr 14, 2023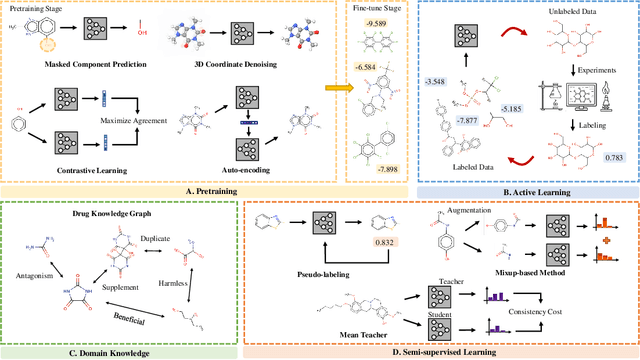
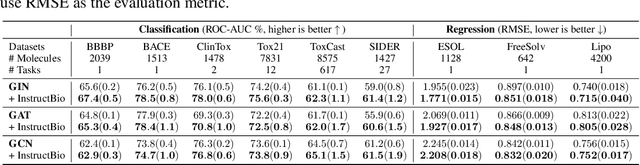
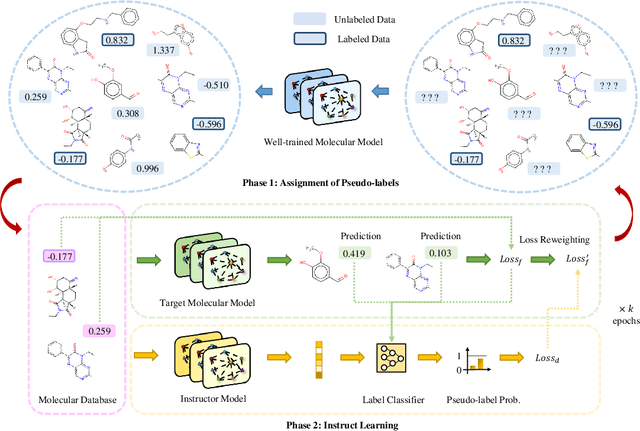
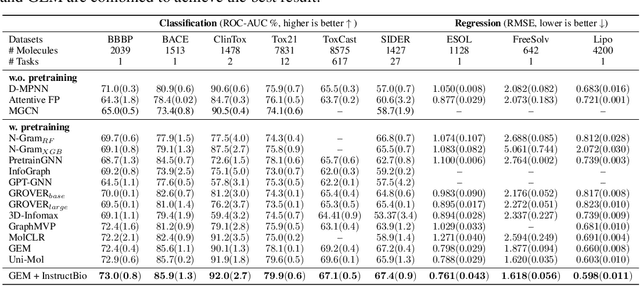
In the field of artificial intelligence for science, it is consistently an essential challenge to face a limited amount of labeled data for real-world problems. The prevailing approach is to pretrain a powerful task-agnostic model on a large unlabeled corpus but may struggle to transfer knowledge to downstream tasks. In this study, we propose InstructMol, a semi-supervised learning algorithm, to take better advantage of unlabeled examples. It introduces an instructor model to provide the confidence ratios as the measurement of pseudo-labels' reliability. These confidence scores then guide the target model to pay distinct attention to different data points, avoiding the over-reliance on labeled data and the negative influence of incorrect pseudo-annotations. Comprehensive experiments show that InstructBio substantially improves the generalization ability of molecular models, in not only molecular property predictions but also activity cliff estimations, demonstrating the superiority of the proposed method. Furthermore, our evidence indicates that InstructBio can be equipped with cutting-edge pretraining methods and used to establish large-scale and task-specific pseudo-labeled molecular datasets, which reduces the predictive errors and shortens the training process. Our work provides strong evidence that semi-supervised learning can be a promising tool to overcome the data scarcity limitation and advance molecular representation learning.
When Geometric Deep Learning Meets Pretrained Protein Language Models
Dec 07, 2022Geometric deep learning has recently achieved great success in non-Euclidean domains, and learning on 3D structures of large biomolecules is emerging as a distinct research area. However, its efficacy is largely constrained due to the limited quantity of structural data. Meanwhile, protein language models trained on substantial 1D sequences have shown burgeoning capabilities with scale in a broad range of applications. Nevertheless, no preceding studies consider combining these different protein modalities to promote the representation power of geometric neural networks. To address this gap, we make the foremost step to integrate the knowledge learned by well-trained protein language models into several state-of-the-art geometric networks. Experiments are evaluated on a variety of protein representation learning benchmarks, including protein-protein interface prediction, model quality assessment, protein-protein rigid-body docking, and binding affinity prediction, leading to an overall improvement of 20% over baselines and the new state-of-the-art performance. Strong evidence indicates that the incorporation of protein language models' knowledge enhances geometric networks' capacity by a significant margin and can be generalized to complex tasks.
Folding membrane proteins by deep transfer learning
Aug 28, 2017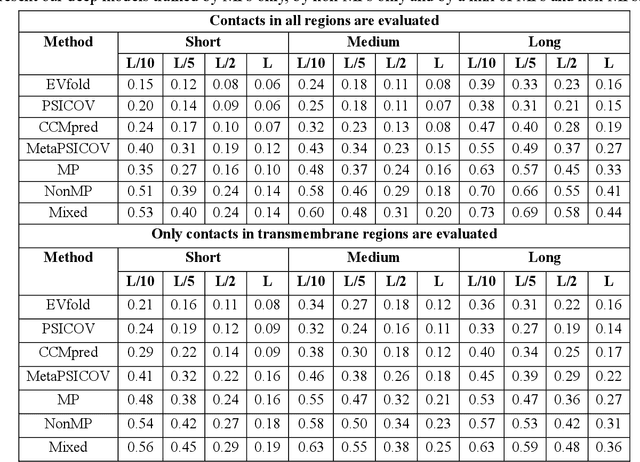
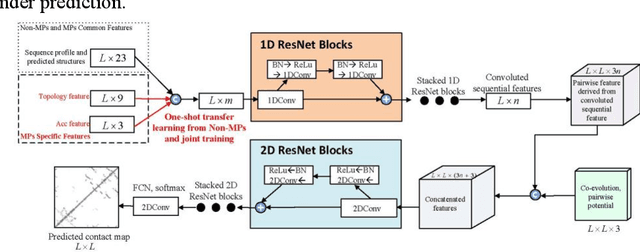
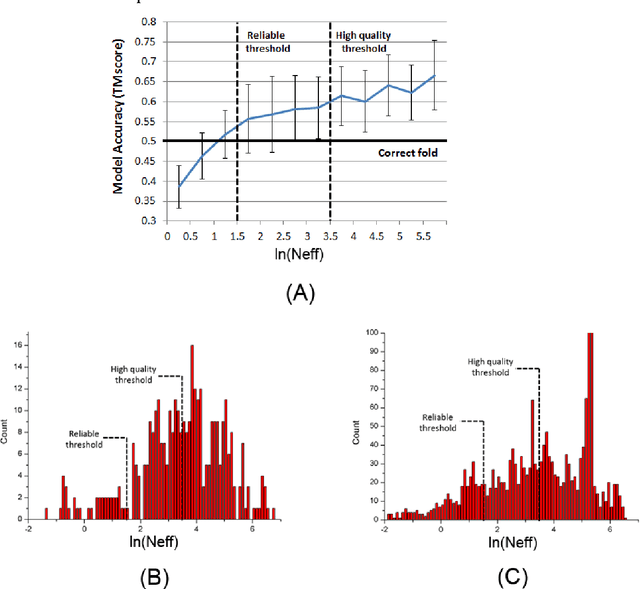
Computational elucidation of membrane protein (MP) structures is challenging partially due to lack of sufficient solved structures for homology modeling. Here we describe a high-throughput deep transfer learning method that first predicts MP contacts by learning from non-membrane proteins (non-MPs) and then predicting three-dimensional structure models using the predicted contacts as distance restraints. Tested on 510 non-redundant MPs, our method has contact prediction accuracy at least 0.18 better than existing methods, predicts correct folds for 218 MPs (TMscore at least 0.6), and generates three-dimensional models with RMSD less than 4 Angstrom and 5 Angstrom for 57 and 108 MPs, respectively. A rigorous blind test in the continuous automated model evaluation (CAMEO) project shows that our method predicted high-resolution three-dimensional models for two recent test MPs of 210 residues with RMSD close to 2 Angstrom. We estimated that our method could predict correct folds for between 1,345 and 1,871 reviewed human multi-pass MPs including a few hundred new folds, which shall facilitate the discovery of drugs targeting at membrane proteins.
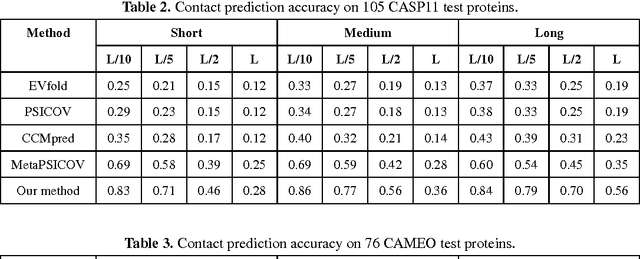
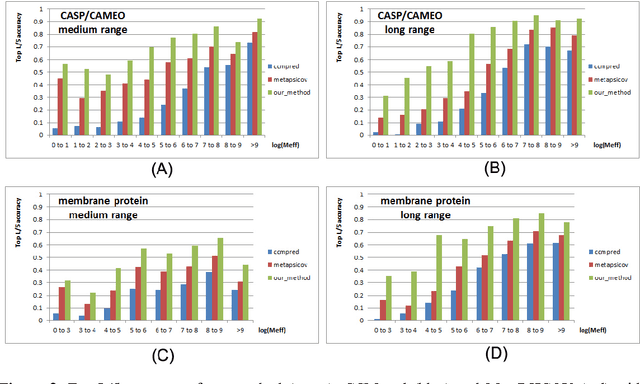
Predicting membrane protein contacts from non-membrane proteins by deep transfer learning
Apr 24, 2017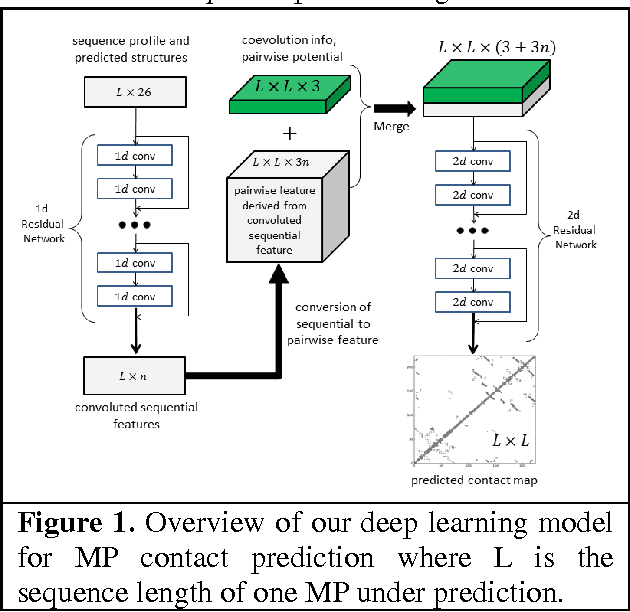
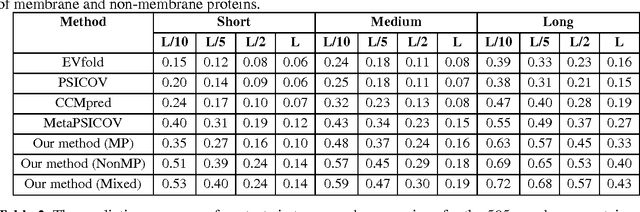
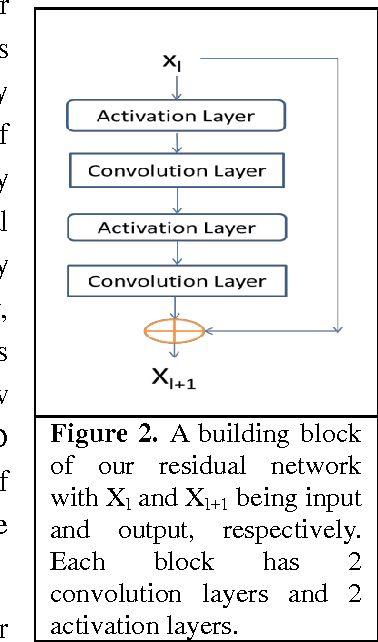
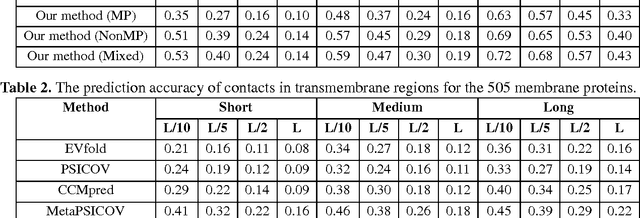
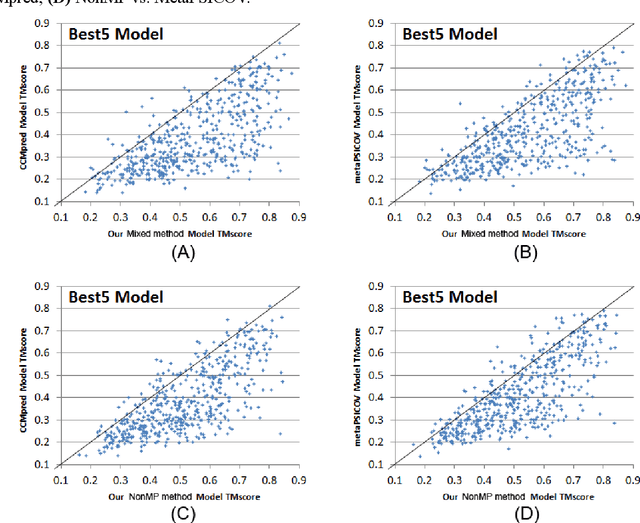
Computational prediction of membrane protein (MP) structures is very challenging partially due to lack of sufficient solved structures for homology modeling. Recently direct evolutionary coupling analysis (DCA) sheds some light on protein contact prediction and accordingly, contact-assisted folding, but DCA is effective only on some very large-sized families since it uses information only in a single protein family. This paper presents a deep transfer learning method that can significantly improve MP contact prediction by learning contact patterns and complex sequence-contact relationship from thousands of non-membrane proteins (non-MPs). Tested on 510 non-redundant MPs, our deep model (learned from only non-MPs) has top L/10 long-range contact prediction accuracy 0.69, better than our deep model trained by only MPs (0.63) and much better than a representative DCA method CCMpred (0.47) and the CASP11 winner MetaPSICOV (0.55). The accuracy of our deep model can be further improved to 0.72 when trained by a mix of non-MPs and MPs. When only contacts in transmembrane regions are evaluated, our method has top L/10 long-range accuracy 0.62, 0.57, and 0.53 when trained by a mix of non-MPs and MPs, by non-MPs only, and by MPs only, respectively, still much better than MetaPSICOV (0.45) and CCMpred (0.40). All these results suggest that sequence-structure relationship learned by our deep model from non-MPs generalizes well to MP contact prediction. Improved contact prediction also leads to better contact-assisted folding. Using only top predicted contacts as restraints, our deep learning method can fold 160 and 200 of 510 MPs with TMscore>0.6 when trained by non-MPs only and by a mix of non-MPs and MPs, respectively, while CCMpred and MetaPSICOV can do so for only 56 and 77 MPs, respectively. Our contact-assisted folding also greatly outperforms homology modeling.
Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model
Nov 27, 2016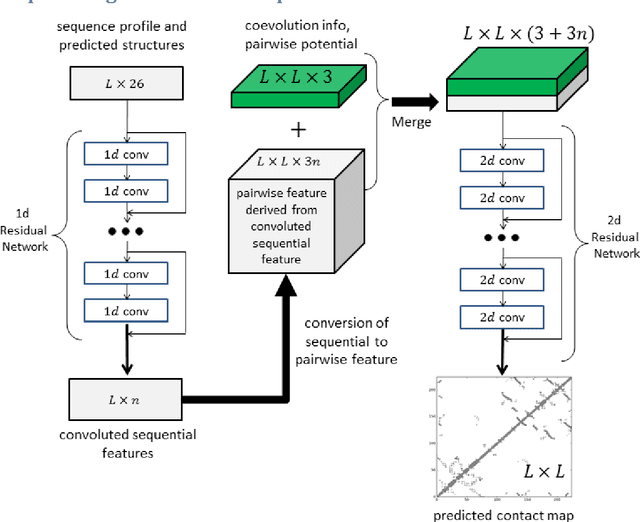
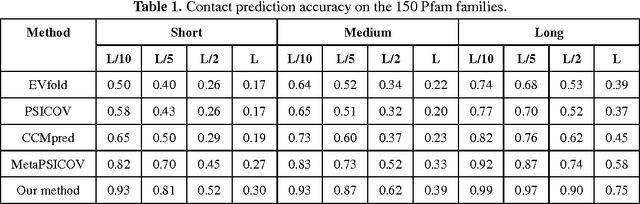
Recently exciting progress has been made on protein contact prediction, but the predicted contacts for proteins without many sequence homologs is still of low quality and not very useful for de novo structure prediction. This paper presents a new deep learning method that predicts contacts by integrating both evolutionary coupling (EC) and sequence conservation information through an ultra-deep neural network formed by two deep residual networks. This deep neural network allows us to model very complex sequence-contact relationship as well as long-range inter-contact correlation. Our method greatly outperforms existing contact prediction methods and leads to much more accurate contact-assisted protein folding. Tested on three datasets of 579 proteins, the average top L long-range prediction accuracy obtained our method, the representative EC method CCMpred and the CASP11 winner MetaPSICOV is 0.47, 0.21 and 0.30, respectively; the average top L/10 long-range accuracy of our method, CCMpred and MetaPSICOV is 0.77, 0.47 and 0.59, respectively. Ab initio folding using our predicted contacts as restraints can yield correct folds (i.e., TMscore>0.6) for 203 test proteins, while that using MetaPSICOV- and CCMpred-predicted contacts can do so for only 79 and 62 proteins, respectively. Further, our contact-assisted models have much better quality than template-based models. Using our predicted contacts as restraints, we can (ab initio) fold 208 of the 398 membrane proteins with TMscore>0.5. By contrast, when the training proteins of our method are used as templates, homology modeling can only do so for 10 of them. One interesting finding is that even if we do not train our prediction models with any membrane proteins, our method works very well on membrane protein prediction. Finally, in recent blind CAMEO benchmark our method successfully folded 5 test proteins with a novel fold.