 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFold-CP: A Context Parallelism Framework for Biomolecular Modeling
Mar 16, 2026Understanding cellular machinery requires atomic-scale reconstruction of large biomolecular assemblies. However, predicting the structures of these systems has been constrained by hardware memory requirements of models like AlphaFold 3, imposing a practical ceiling of a few thousand residues that can be processed on a single GPU. Here we present NVIDIA BioNeMo Fold-CP, a context parallelism framework that overcomes this barrier by distributing the inference and training pipelines of co-folding models across multiple GPUs. We use the Boltz models as open source reference architectures and implement custom multidimensional primitives that efficiently parallelize both the dense triangular updates and the irregular, data-dependent pattern of window-batched local attention. Our approach achieves efficient memory scaling; for an N-token input distributed across P GPUs, per-device memory scales as $O(N^2/P)$, enabling the structure prediction of assemblies exceeding 30,000 residues on 64 NVIDIA B300 GPUs. We demonstrate the scientific utility of this approach through successful developer use cases: Fold-CP enabled the scoring of over 90% of Comprehensive Resource of Mammalian protein complexes (CORUM) database, as well as folding of disease-relevant PI4KA lipid kinase complex bound to an intrinsically disordered region without cropping. By providing a scalable pathway for modeling massive systems with full global context, Fold-CP represents a significant step toward the realization of a virtual cell.
ESNERA: Empirical and semantic named entity alignment for named entity dataset merging
Aug 09, 2025Named Entity Recognition (NER) is a fundamental task in natural language processing. It remains a research hotspot due to its wide applicability across domains. Although recent advances in deep learning have significantly improved NER performance, they rely heavily on large, high-quality annotated datasets. However, building these datasets is expensive and time-consuming, posing a major bottleneck for further research. Current dataset merging approaches mainly focus on strategies like manual label mapping or constructing label graphs, which lack interpretability and scalability. To address this, we propose an automatic label alignment method based on label similarity. The method combines empirical and semantic similarities, using a greedy pairwise merging strategy to unify label spaces across different datasets. Experiments are conducted in two stages: first, merging three existing NER datasets into a unified corpus with minimal impact on NER performance; second, integrating this corpus with a small-scale, self-built dataset in the financial domain. The results show that our method enables effective dataset merging and enhances NER performance in the low-resource financial domain. This study presents an efficient, interpretable, and scalable solution for integrating multi-source NER corpora.
Skywork-R1V3 Technical Report
Jul 09, 2025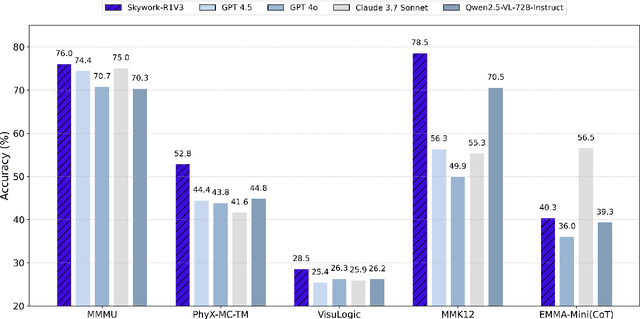
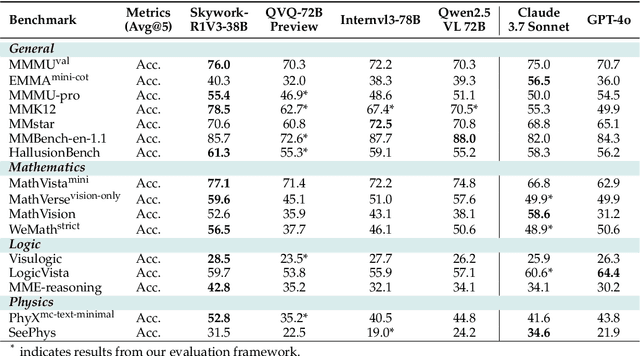
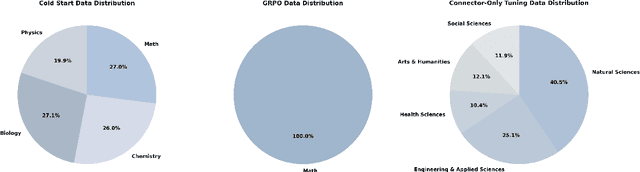
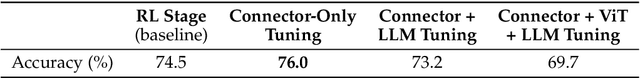
We introduce Skywork-R1V3, an advanced, open-source vision-language model (VLM) that pioneers a new approach to visual reasoning. Its key innovation lies in effectively transferring reasoning skills from text-only Large Language Models (LLMs) to visual tasks. The strong performance of Skywork-R1V3 primarily stems from our elaborate post-training RL framework, which effectively activates and enhances the model's reasoning ability, without the need for additional continue pre-training. Through this framework, we further uncover the fundamental role of the connector module in achieving robust cross-modal alignment for multimodal reasoning models. In addition, we introduce a unique indicator of reasoning capability, the entropy of critical reasoning tokens, which has proven highly effective for checkpoint selection during RL training. Skywork-R1V3 achieves state-of-the-art results on MMMU, significantly improving from 64.3% to 76.0%. This performance matches entry-level human capabilities. Remarkably, our RL-powered post-training approach enables even the 38B parameter model to rival top closed-source VLMs. The implementation successfully transfers mathematical reasoning to other subject-related reasoning tasks. We also include an analysis of curriculum learning and reinforcement finetuning strategies, along with a broader discussion on multimodal reasoning. Skywork-R1V3 represents a significant leap in multimodal reasoning, showcasing RL as a powerful engine for advancing open-source VLM capabilities.
SpNeRF: Memory Efficient Sparse Volumetric Neural Rendering Accelerator for Edge Devices
May 13, 2025Neural rendering has gained prominence for its high-quality output, which is crucial for AR/VR applications. However, its large voxel grid data size and irregular access patterns challenge real-time processing on edge devices. While previous works have focused on improving data locality, they have not adequately addressed the issue of large voxel grid sizes, which necessitate frequent off-chip memory access and substantial on-chip memory. This paper introduces SpNeRF, a software-hardware co-design solution tailored for sparse volumetric neural rendering. We first identify memory-bound rendering inefficiencies and analyze the inherent sparsity in the voxel grid data of neural rendering. To enhance efficiency, we propose novel preprocessing and online decoding steps, reducing the memory size for voxel grid. The preprocessing step employs hash mapping to support irregular data access while maintaining a minimal memory size. The online decoding step enables efficient on-chip sparse voxel grid processing, incorporating bitmap masking to mitigate PSNR loss caused by hash collisions. To further optimize performance, we design a dedicated hardware architecture supporting our sparse voxel grid processing technique. Experimental results demonstrate that SpNeRF achieves an average 21.07$\times$ reduction in memory size while maintaining comparable PSNR levels. When benchmarked against Jetson XNX, Jetson ONX, RT-NeRF.Edge and NeuRex.Edge, our design achieves speedups of 95.1$\times$, 63.5$\times$, 1.5$\times$ and 10.3$\times$, and improves energy efficiency by 625.6$\times$, 529.1$\times$, 4$\times$, and 4.4$\times$, respectively.
DeepForest: Sensing Into Self-Occluding Volumes of Vegetation With Aerial Imaging
Feb 04, 2025



Access to below-canopy volumetric vegetation data is crucial for understanding ecosystem dynamics. We address the long-standing limitation of remote sensing to penetrate deep into dense canopy layers. LiDAR and radar are currently considered the primary options for measuring 3D vegetation structures, while cameras can only extract the reflectance and depth of top layers. Using conventional, high-resolution aerial images, our approach allows sensing deep into self-occluding vegetation volumes, such as forests. It is similar in spirit to the imaging process of wide-field microscopy, but can handle much larger scales and strong occlusion. We scan focal stacks by synthetic-aperture imaging with drones and reduce out-of-focus signal contributions using pre-trained 3D convolutional neural networks. The resulting volumetric reflectance stacks contain low-frequency representations of the vegetation volume. Combining multiple reflectance stacks from various spectral channels provides insights into plant health, growth, and environmental conditions throughout the entire vegetation volume.
Group Ligands Docking to Protein Pockets
Jan 25, 2025



Molecular docking is a key task in computational biology that has attracted increasing interest from the machine learning community. While existing methods have achieved success, they generally treat each protein-ligand pair in isolation. Inspired by the biochemical observation that ligands binding to the same target protein tend to adopt similar poses, we propose \textsc{GroupBind}, a novel molecular docking framework that simultaneously considers multiple ligands docking to a protein. This is achieved by introducing an interaction layer for the group of ligands and a triangle attention module for embedding protein-ligand and group-ligand pairs. By integrating our approach with diffusion-based docking model, we set a new S performance on the PDBBind blind docking benchmark, demonstrating the effectiveness of our proposed molecular docking paradigm.
Hotspot-Driven Peptide Design via Multi-Fragment Autoregressive Extension
Nov 26, 2024



Peptides, short chains of amino acids, interact with target proteins, making them a unique class of protein-based therapeutics for treating human diseases. Recently, deep generative models have shown great promise in peptide generation. However, several challenges remain in designing effective peptide binders. First, not all residues contribute equally to peptide-target interactions. Second, the generated peptides must adopt valid geometries due to the constraints of peptide bonds. Third, realistic tasks for peptide drug development are still lacking. To address these challenges, we introduce PepHAR, a hot-spot-driven autoregressive generative model for designing peptides targeting specific proteins. Building on the observation that certain hot spot residues have higher interaction potentials, we first use an energy-based density model to fit and sample these key residues. Next, to ensure proper peptide geometry, we autoregressively extend peptide fragments by estimating dihedral angles between residue frames. Finally, we apply an optimization process to iteratively refine fragment assembly, ensuring correct peptide structures. By combining hot spot sampling with fragment-based extension, our approach enables de novo peptide design tailored to a target protein and allows the incorporation of key hot spot residues into peptide scaffolds. Extensive experiments, including peptide design and peptide scaffold generation, demonstrate the strong potential of PepHAR in computational peptide binder design.
GOPT: Generalizable Online 3D Bin Packing via Transformer-based Deep Reinforcement Learning
Sep 09, 2024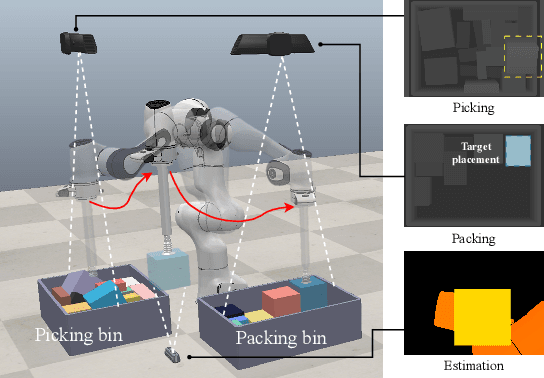
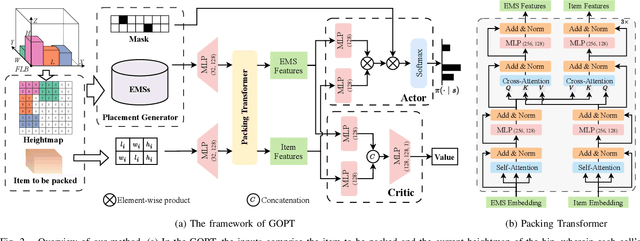
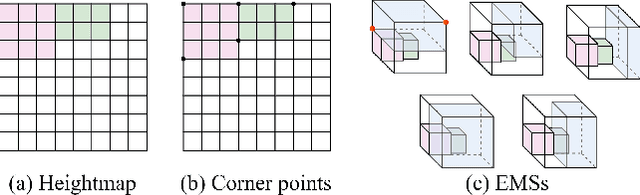
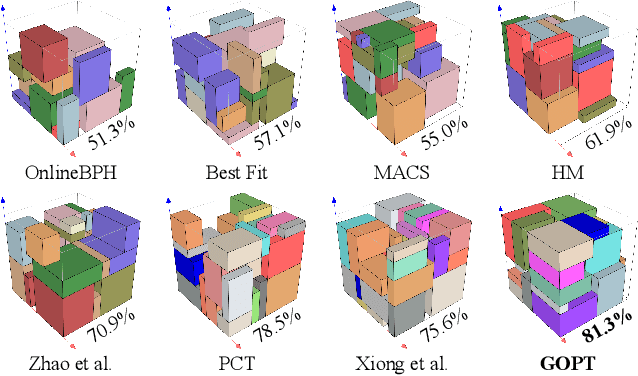
Robotic object packing has broad practical applications in the logistics and automation industry, often formulated by researchers as the online 3D Bin Packing Problem (3D-BPP). However, existing DRL-based methods primarily focus on enhancing performance in limited packing environments while neglecting the ability to generalize across multiple environments characterized by different bin dimensions. To this end, we propose GOPT, a generalizable online 3D Bin Packing approach via Transformer-based deep reinforcement learning (DRL). First, we design a Placement Generator module to yield finite subspaces as placement candidates and the representation of the bin. Second, we propose a Packing Transformer, which fuses the features of the items and bin, to identify the spatial correlation between the item to be packed and available sub-spaces within the bin. Coupling these two components enables GOPT's ability to perform inference on bins of varying dimensions. We conduct extensive experiments and demonstrate that GOPT not only achieves superior performance against the baselines, but also exhibits excellent generalization capabilities. Furthermore, the deployment with a robot showcases the practical applicability of our method in the real world. The source code will be publicly available at https://github.com/Xiong5Heng/GOPT.
FAFE: Immune Complex Modeling with Geodesic Distance Loss on Noisy Group Frames
Jul 01, 2024



Despite the striking success of general protein folding models such as AlphaFold2(AF2, Jumper et al. (2021)), the accurate computational modeling of antibody-antigen complexes remains a challenging task. In this paper, we first analyze AF2's primary loss function, known as the Frame Aligned Point Error (FAPE), and raise a previously overlooked issue that FAPE tends to face gradient vanishing problem on high-rotational-error targets. To address this fundamental limitation, we propose a novel geodesic loss called Frame Aligned Frame Error (FAFE, denoted as F2E to distinguish from FAPE), which enables the model to better optimize both the rotational and translational errors between two frames. We then prove that F2E can be reformulated as a group-aware geodesic loss, which translates the optimization of the residue-to-residue error to optimizing group-to-group geodesic frame distance. By fine-tuning AF2 with our proposed new loss function, we attain a correct rate of 52.3\% (DockQ $>$ 0.23) on an evaluation set and 43.8\% correct rate on a subset with low homology, with substantial improvement over AF2 by 182\% and 100\% respectively.
Full-Atom Peptide Design based on Multi-modal Flow Matching
Jun 02, 2024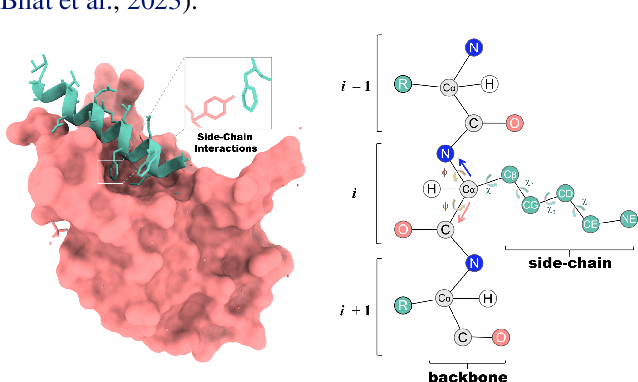
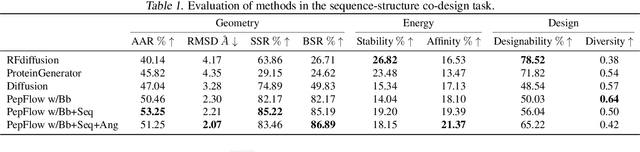
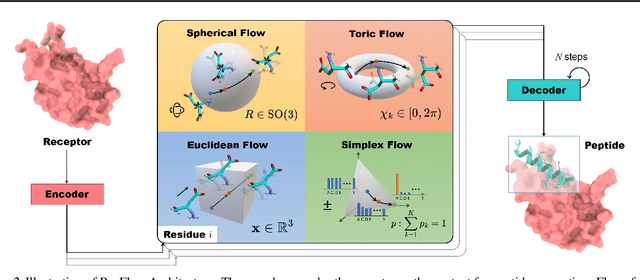
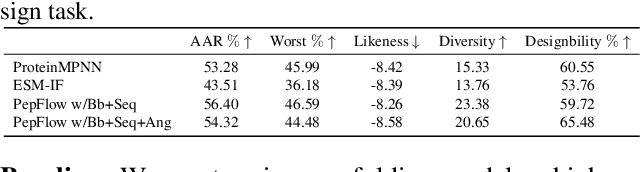
Peptides, short chains of amino acid residues, play a vital role in numerous biological processes by interacting with other target molecules, offering substantial potential in drug discovery. In this work, we present PepFlow, the first multi-modal deep generative model grounded in the flow-matching framework for the design of full-atom peptides that target specific protein receptors. Drawing inspiration from the crucial roles of residue backbone orientations and side-chain dynamics in protein-peptide interactions, we characterize the peptide structure using rigid backbone frames within the $\mathrm{SE}(3)$ manifold and side-chain angles on high-dimensional tori. Furthermore, we represent discrete residue types in the peptide sequence as categorical distributions on the probability simplex. By learning the joint distributions of each modality using derived flows and vector fields on corresponding manifolds, our method excels in the fine-grained design of full-atom peptides. Harnessing the multi-modal paradigm, our approach adeptly tackles various tasks such as fix-backbone sequence design and side-chain packing through partial sampling. Through meticulously crafted experiments, we demonstrate that PepFlow exhibits superior performance in comprehensive benchmarks, highlighting its significant potential in computational peptide design and analysis.