 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePeptide2Mol: A Diffusion Model for Generating Small Molecules as Peptide Mimics for Targeted Protein Binding
Nov 07, 2025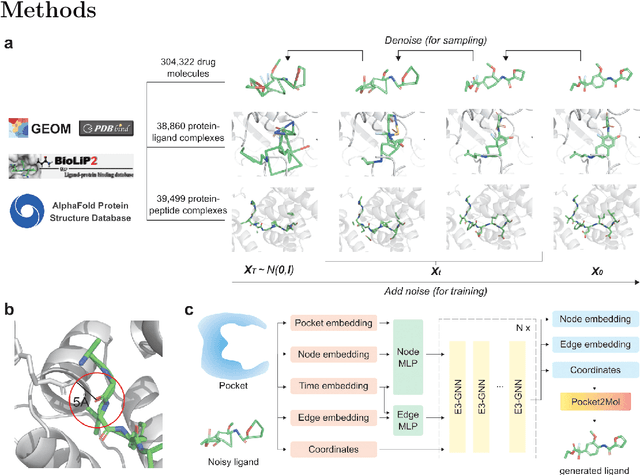
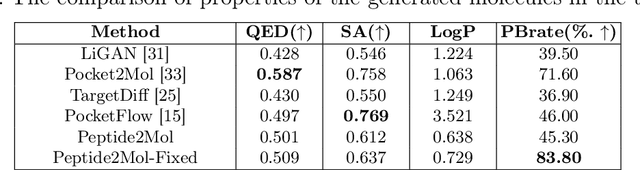
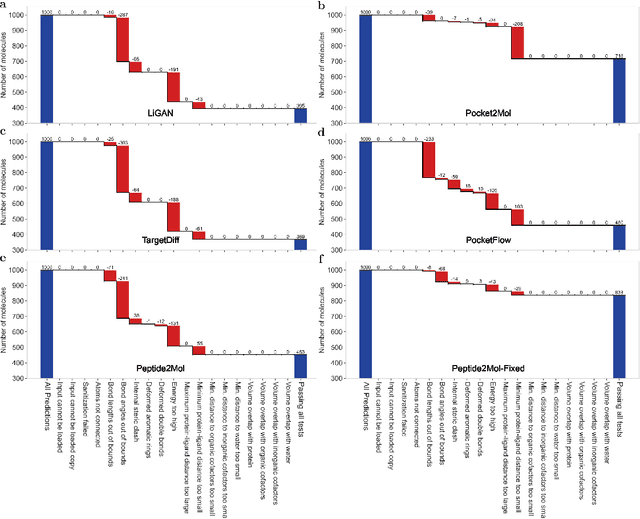
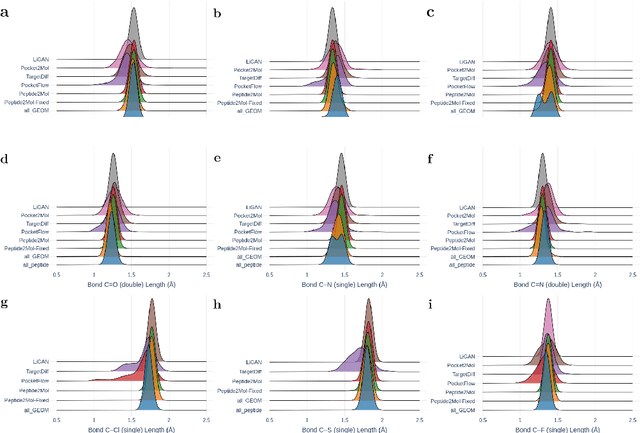
Structure-based drug design has seen significant advancements with the integration of artificial intelligence (AI), particularly in the generation of hit and lead compounds. However, most AI-driven approaches neglect the importance of endogenous protein interactions with peptides, which may result in suboptimal molecule designs. In this work, we present Peptide2Mol, an E(3)-equivariant graph neural network diffusion model that generates small molecules by referencing both the original peptide binders and their surrounding protein pocket environments. Trained on large datasets and leveraging sophisticated modeling techniques, Peptide2Mol not only achieves state-of-the-art performance in non-autoregressive generative tasks, but also produces molecules with similarity to the original peptide binder. Additionally, the model allows for molecule optimization and peptidomimetic design through a partial diffusion process. Our results highlight Peptide2Mol as an effective deep generative model for generating and optimizing bioactive small molecules from protein binding pockets.
Group Ligands Docking to Protein Pockets
Jan 25, 2025



Molecular docking is a key task in computational biology that has attracted increasing interest from the machine learning community. While existing methods have achieved success, they generally treat each protein-ligand pair in isolation. Inspired by the biochemical observation that ligands binding to the same target protein tend to adopt similar poses, we propose \textsc{GroupBind}, a novel molecular docking framework that simultaneously considers multiple ligands docking to a protein. This is achieved by introducing an interaction layer for the group of ligands and a triangle attention module for embedding protein-ligand and group-ligand pairs. By integrating our approach with diffusion-based docking model, we set a new S performance on the PDBBind blind docking benchmark, demonstrating the effectiveness of our proposed molecular docking paradigm.
Reprogramming Pretrained Target-Specific Diffusion Models for Dual-Target Drug Design
Oct 28, 2024


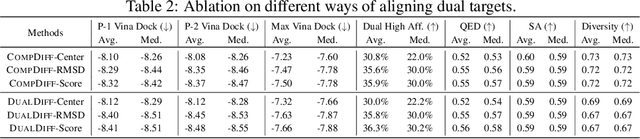
Dual-target therapeutic strategies have become a compelling approach and attracted significant attention due to various benefits, such as their potential in overcoming drug resistance in cancer therapy. Considering the tremendous success that deep generative models have achieved in structure-based drug design in recent years, we formulate dual-target drug design as a generative task and curate a novel dataset of potential target pairs based on synergistic drug combinations. We propose to design dual-target drugs with diffusion models that are trained on single-target protein-ligand complex pairs. Specifically, we align two pockets in 3D space with protein-ligand binding priors and build two complex graphs with shared ligand nodes for SE(3)-equivariant composed message passing, based on which we derive a composed drift in both 3D and categorical probability space in the generative process. Our algorithm can well transfer the knowledge gained in single-target pretraining to dual-target scenarios in a zero-shot manner. We also repurpose linker design methods as strong baselines for this task. Extensive experiments demonstrate the effectiveness of our method compared with various baselines.
Geometric Representation Condition Improves Equivariant Molecule Generation
Oct 04, 2024



Recent advancements in molecular generative models have demonstrated substantial potential in accelerating scientific discovery, particularly in drug design. However, these models often face challenges in generating high-quality molecules, especially in conditional scenarios where specific molecular properties must be satisfied. In this work, we introduce GeoRCG, a general framework to enhance the performance of molecular generative models by integrating geometric representation conditions. We decompose the molecule generation process into two stages: first, generating an informative geometric representation; second, generating a molecule conditioned on the representation. Compared to directly generating a molecule, the relatively easy-to-generate representation in the first-stage guides the second-stage generation to reach a high-quality molecule in a more goal-oriented and much faster way. Leveraging EDM as the base generator, we observe significant quality improvements in unconditional molecule generation on the widely-used QM9 and GEOM-DRUG datasets. More notably, in the challenging conditional molecular generation task, our framework achieves an average 31\% performance improvement over state-of-the-art approaches, highlighting the superiority of conditioning on semantically rich geometric representations over conditioning on individual property values as in previous approaches. Furthermore, we show that, with such representation guidance, the number of diffusion steps can be reduced to as small as 100 while maintaining superior generation quality than that achieved with 1,000 steps, thereby significantly accelerating the generation process.
MolDiff: Addressing the Atom-Bond Inconsistency Problem in 3D Molecule Diffusion Generation
May 11, 2023Deep generative models have recently achieved superior performance in 3D molecule generation. Most of them first generate atoms and then add chemical bonds based on the generated atoms in a post-processing manner. However, there might be no corresponding bond solution for the temporally generated atoms as their locations are generated without considering potential bonds. We define this problem as the atom-bond inconsistency problem and claim it is the main reason for current approaches to generating unrealistic 3D molecules. To overcome this problem, we propose a new diffusion model called MolDiff which can generate atoms and bonds simultaneously while still maintaining their consistency by explicitly modeling the dependence between their relationships. We evaluated the generation ability of our proposed model and the quality of the generated molecules using criteria related to both geometry and chemical properties. The empirical studies showed that our model outperforms previous approaches, achieving a three-fold improvement in success rate and generating molecules with significantly better quality.
3D Equivariant Diffusion for Target-Aware Molecule Generation and Affinity Prediction
Mar 06, 2023



Rich data and powerful machine learning models allow us to design drugs for a specific protein target \textit{in silico}. Recently, the inclusion of 3D structures during targeted drug design shows superior performance to other target-free models as the atomic interaction in the 3D space is explicitly modeled. However, current 3D target-aware models either rely on the voxelized atom densities or the autoregressive sampling process, which are not equivariant to rotation or easily violate geometric constraints resulting in unrealistic structures. In this work, we develop a 3D equivariant diffusion model to solve the above challenges. To achieve target-aware molecule design, our method learns a joint generative process of both continuous atom coordinates and categorical atom types with a SE(3)-equivariant network. Moreover, we show that our model can serve as an unsupervised feature extractor to estimate the binding affinity under proper parameterization, which provides an effective way for drug screening. To evaluate our model, we propose a comprehensive framework to evaluate the quality of sampled molecules from different dimensions. Empirical studies show our model could generate molecules with more realistic 3D structures and better affinities towards the protein targets, and improve binding affinity ranking and prediction without retraining.
3DLinker: An E(3) Equivariant Variational Autoencoder for Molecular Linker Design
May 15, 2022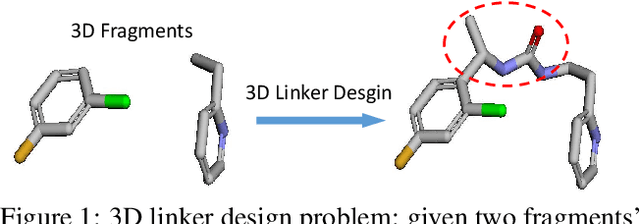
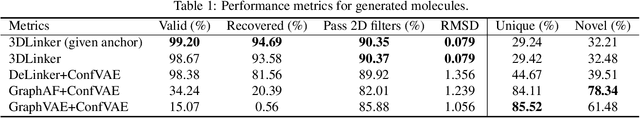
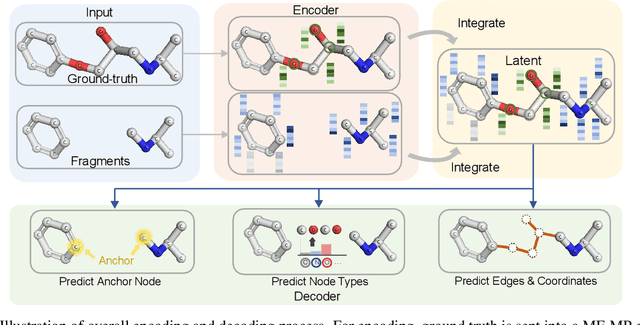
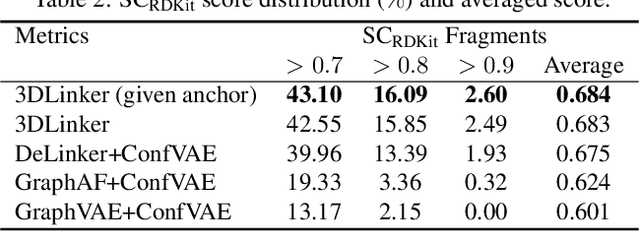
Deep learning has achieved tremendous success in designing novel chemical compounds with desirable pharmaceutical properties. In this work, we focus on a new type of drug design problem -- generating a small "linker" to physically attach two independent molecules with their distinct functions. The main computational challenges include: 1) the generation of linkers is conditional on the two given molecules, in contrast to generating full molecules from scratch in previous works; 2) linkers heavily depend on the anchor atoms of the two molecules to be connected, which are not known beforehand; 3) 3D structures and orientations of the molecules need to be considered to avoid atom clashes, for which equivariance to E(3) group are necessary. To address these problems, we propose a conditional generative model, named 3DLinker, which is able to predict anchor atoms and jointly generate linker graphs and their 3D structures based on an E(3) equivariant graph variational autoencoder. So far as we know, there are no previous models that could achieve this task. We compare our model with multiple conditional generative models modified from other molecular design tasks and find that our model has a significantly higher rate in recovering molecular graphs, and more importantly, accurately predicting the 3D coordinates of all the atoms.
Pocket2Mol: Efficient Molecular Sampling Based on 3D Protein Pockets
May 15, 2022



Deep generative models have achieved tremendous success in designing novel drug molecules in recent years. A new thread of works have shown the great potential in advancing the specificity and success rate of in silico drug design by considering the structure of protein pockets. This setting posts fundamental computational challenges in sampling new chemical compounds that could satisfy multiple geometrical constraints imposed by pockets. Previous sampling algorithms either sample in the graph space or only consider the 3D coordinates of atoms while ignoring other detailed chemical structures such as bond types and functional groups. To address the challenge, we develop Pocket2Mol, an E(3)-equivariant generative network composed of two modules: 1) a new graph neural network capturing both spatial and bonding relationships between atoms of the binding pockets and 2) a new efficient algorithm which samples new drug candidates conditioned on the pocket representations from a tractable distribution without relying on MCMC. Experimental results demonstrate that molecules sampled from Pocket2Mol achieve significantly better binding affinity and other drug properties such as druglikeness and synthetic accessibility.