 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHotspot-Driven Peptide Design via Multi-Fragment Autoregressive Extension
Nov 26, 2024



Peptides, short chains of amino acids, interact with target proteins, making them a unique class of protein-based therapeutics for treating human diseases. Recently, deep generative models have shown great promise in peptide generation. However, several challenges remain in designing effective peptide binders. First, not all residues contribute equally to peptide-target interactions. Second, the generated peptides must adopt valid geometries due to the constraints of peptide bonds. Third, realistic tasks for peptide drug development are still lacking. To address these challenges, we introduce PepHAR, a hot-spot-driven autoregressive generative model for designing peptides targeting specific proteins. Building on the observation that certain hot spot residues have higher interaction potentials, we first use an energy-based density model to fit and sample these key residues. Next, to ensure proper peptide geometry, we autoregressively extend peptide fragments by estimating dihedral angles between residue frames. Finally, we apply an optimization process to iteratively refine fragment assembly, ensuring correct peptide structures. By combining hot spot sampling with fragment-based extension, our approach enables de novo peptide design tailored to a target protein and allows the incorporation of key hot spot residues into peptide scaffolds. Extensive experiments, including peptide design and peptide scaffold generation, demonstrate the strong potential of PepHAR in computational peptide binder design.
FAFE: Immune Complex Modeling with Geodesic Distance Loss on Noisy Group Frames
Jul 01, 2024



Despite the striking success of general protein folding models such as AlphaFold2(AF2, Jumper et al. (2021)), the accurate computational modeling of antibody-antigen complexes remains a challenging task. In this paper, we first analyze AF2's primary loss function, known as the Frame Aligned Point Error (FAPE), and raise a previously overlooked issue that FAPE tends to face gradient vanishing problem on high-rotational-error targets. To address this fundamental limitation, we propose a novel geodesic loss called Frame Aligned Frame Error (FAFE, denoted as F2E to distinguish from FAPE), which enables the model to better optimize both the rotational and translational errors between two frames. We then prove that F2E can be reformulated as a group-aware geodesic loss, which translates the optimization of the residue-to-residue error to optimizing group-to-group geodesic frame distance. By fine-tuning AF2 with our proposed new loss function, we attain a correct rate of 52.3\% (DockQ $>$ 0.23) on an evaluation set and 43.8\% correct rate on a subset with low homology, with substantial improvement over AF2 by 182\% and 100\% respectively.
Full-Atom Peptide Design based on Multi-modal Flow Matching
Jun 02, 2024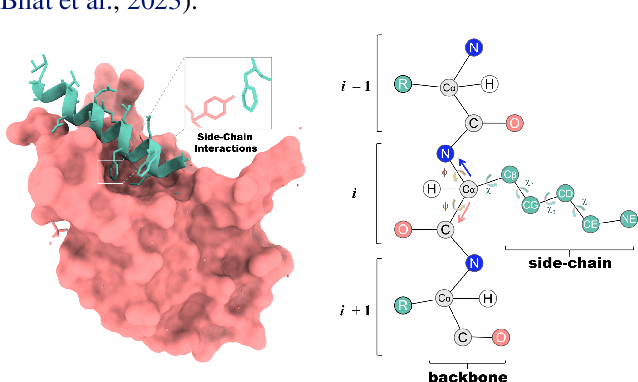
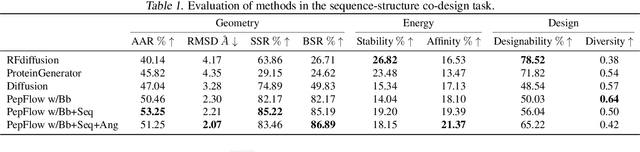
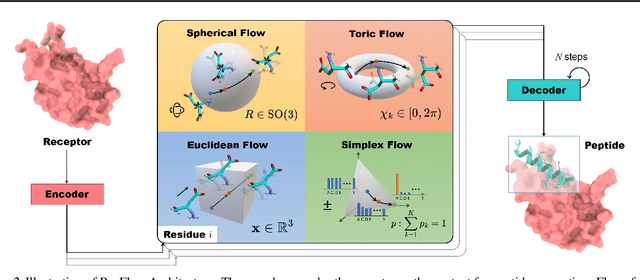
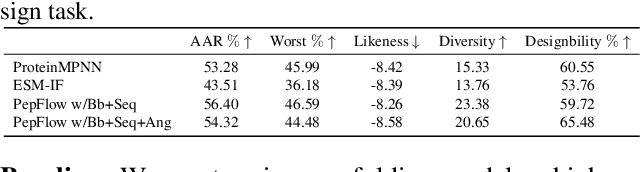
Peptides, short chains of amino acid residues, play a vital role in numerous biological processes by interacting with other target molecules, offering substantial potential in drug discovery. In this work, we present PepFlow, the first multi-modal deep generative model grounded in the flow-matching framework for the design of full-atom peptides that target specific protein receptors. Drawing inspiration from the crucial roles of residue backbone orientations and side-chain dynamics in protein-peptide interactions, we characterize the peptide structure using rigid backbone frames within the $\mathrm{SE}(3)$ manifold and side-chain angles on high-dimensional tori. Furthermore, we represent discrete residue types in the peptide sequence as categorical distributions on the probability simplex. By learning the joint distributions of each modality using derived flows and vector fields on corresponding manifolds, our method excels in the fine-grained design of full-atom peptides. Harnessing the multi-modal paradigm, our approach adeptly tackles various tasks such as fix-backbone sequence design and side-chain packing through partial sampling. Through meticulously crafted experiments, we demonstrate that PepFlow exhibits superior performance in comprehensive benchmarks, highlighting its significant potential in computational peptide design and analysis.
Pocket2Mol: Efficient Molecular Sampling Based on 3D Protein Pockets
May 15, 2022



Deep generative models have achieved tremendous success in designing novel drug molecules in recent years. A new thread of works have shown the great potential in advancing the specificity and success rate of in silico drug design by considering the structure of protein pockets. This setting posts fundamental computational challenges in sampling new chemical compounds that could satisfy multiple geometrical constraints imposed by pockets. Previous sampling algorithms either sample in the graph space or only consider the 3D coordinates of atoms while ignoring other detailed chemical structures such as bond types and functional groups. To address the challenge, we develop Pocket2Mol, an E(3)-equivariant generative network composed of two modules: 1) a new graph neural network capturing both spatial and bonding relationships between atoms of the binding pockets and 2) a new efficient algorithm which samples new drug candidates conditioned on the pocket representations from a tractable distribution without relying on MCMC. Experimental results demonstrate that molecules sampled from Pocket2Mol achieve significantly better binding affinity and other drug properties such as druglikeness and synthetic accessibility.
Equivariant Point Cloud Analysis via Learning Orientations for Message Passing
Mar 28, 2022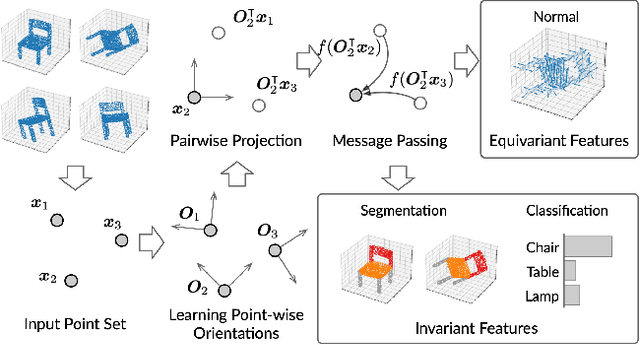
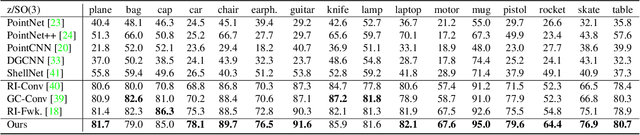
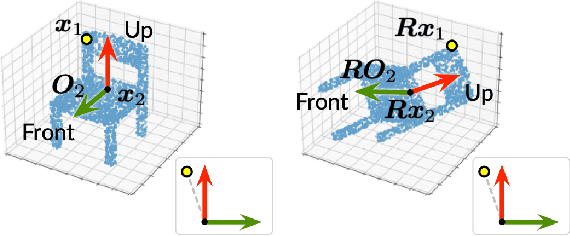
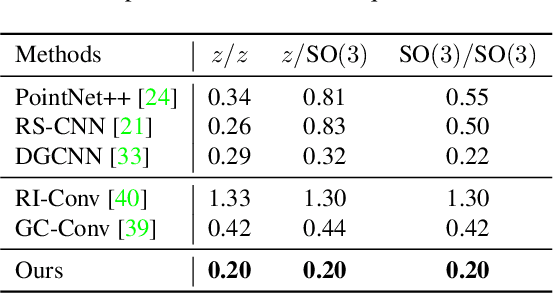
Equivariance has been a long-standing concern in various fields ranging from computer vision to physical modeling. Most previous methods struggle with generality, simplicity, and expressiveness -- some are designed ad hoc for specific data types, some are too complex to be accessible, and some sacrifice flexible transformations. In this work, we propose a novel and simple framework to achieve equivariance for point cloud analysis based on the message passing (graph neural network) scheme. We find the equivariant property could be obtained by introducing an orientation for each point to decouple the relative position for each point from the global pose of the entire point cloud. Therefore, we extend current message passing networks with a module that learns orientations for each point. Before aggregating information from the neighbors of a point, the networks transforms the neighbors' coordinates based on the point's learned orientations. We provide formal proofs to show the equivariance of the proposed framework. Empirically, we demonstrate that our proposed method is competitive on both point cloud analysis and physical modeling tasks. Code is available at https://github.com/luost26/Equivariant-OrientedMP .
A 3D Molecule Generative Model for Structure-Based Drug Design
Mar 20, 2022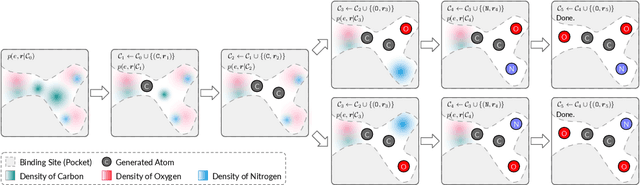
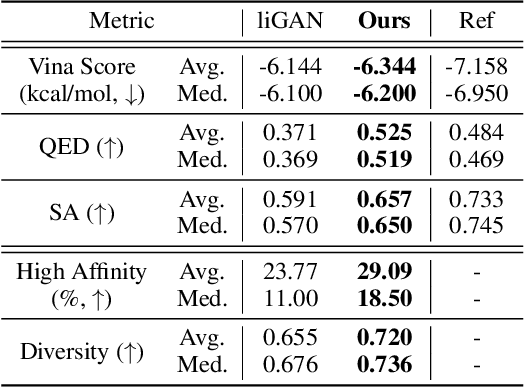
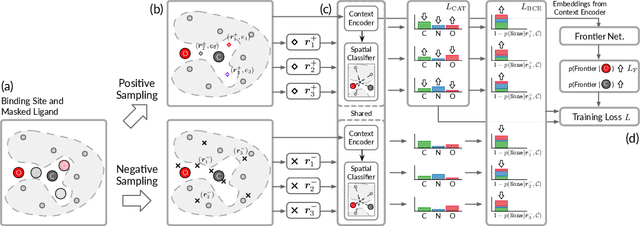
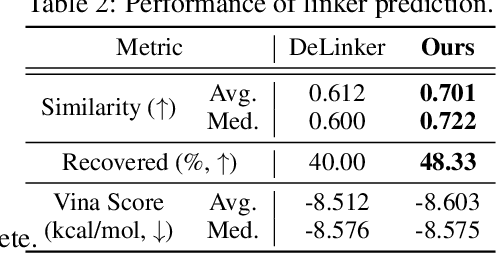
We study a fundamental problem in structure-based drug design -- generating molecules that bind to specific protein binding sites. While we have witnessed the great success of deep generative models in drug design, the existing methods are mostly string-based or graph-based. They are limited by the lack of spatial information and thus unable to be applied to structure-based design tasks. Particularly, such models have no or little knowledge of how molecules interact with their target proteins exactly in 3D space. In this paper, we propose a 3D generative model that generates molecules given a designated 3D protein binding site. Specifically, given a binding site as the 3D context, our model estimates the probability density of atom's occurrences in 3D space -- positions that are more likely to have atoms will be assigned higher probability. To generate 3D molecules, we propose an auto-regressive sampling scheme -- atoms are sampled sequentially from the learned distribution until there is no room for new atoms. Combined with this sampling scheme, our model can generate valid and diverse molecules, which could be applicable to various structure-based molecular design tasks such as molecule sampling and linker design. Experimental results demonstrate that molecules sampled from our model exhibit high binding affinity to specific targets and good drug properties such as drug-likeness even if the model is not explicitly optimized for them.
Directed Weight Neural Networks for Protein Structure Representation Learning
Jan 28, 2022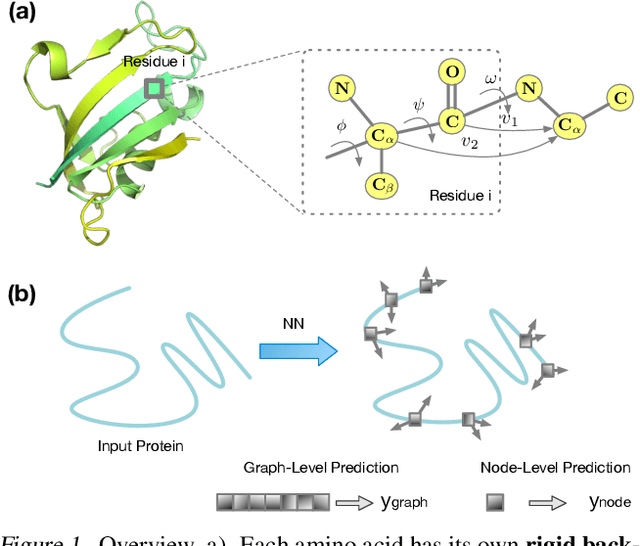
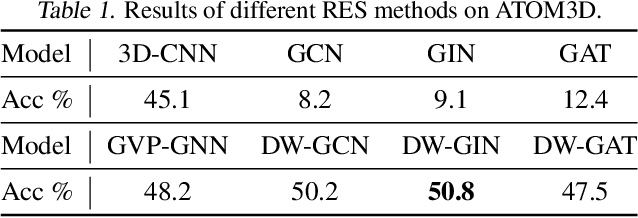
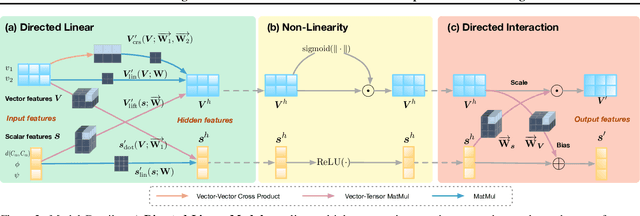
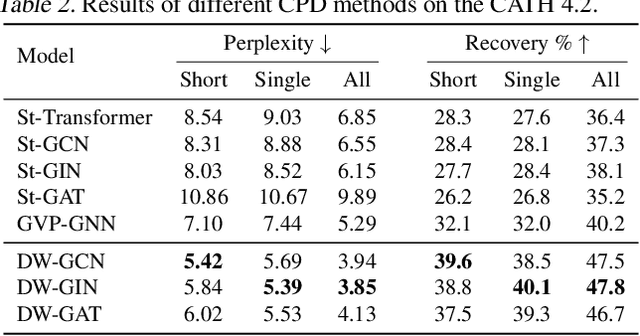
A protein performs biological functions by folding to a particular 3D structure. To accurately model the protein structures, both the overall geometric topology and local fine-grained relations between amino acids (e.g. side-chain torsion angles and inter-amino-acid orientations) should be carefully considered. In this work, we propose the Directed Weight Neural Network for better capturing geometric relations among different amino acids. Extending a single weight from a scalar to a 3D directed vector, our new framework supports a rich set of geometric operations on both classical and SO(3)--representation features, on top of which we construct a perceptron unit for processing amino-acid information. In addition, we introduce an equivariant message passing paradigm on proteins for plugging the directed weight perceptrons into existing Graph Neural Networks, showing superior versatility in maintaining SO(3)-equivariance at the global scale. Experiments show that our network has remarkably better expressiveness in representing geometric relations in comparison to classical neural networks and the (globally) equivariant networks. It also achieves state-of-the-art performance on various computational biology applications related to protein 3D structures.
Deep Point Set Resampling via Gradient Fields
Nov 03, 2021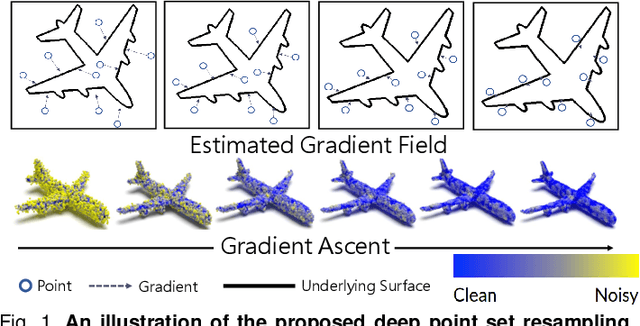
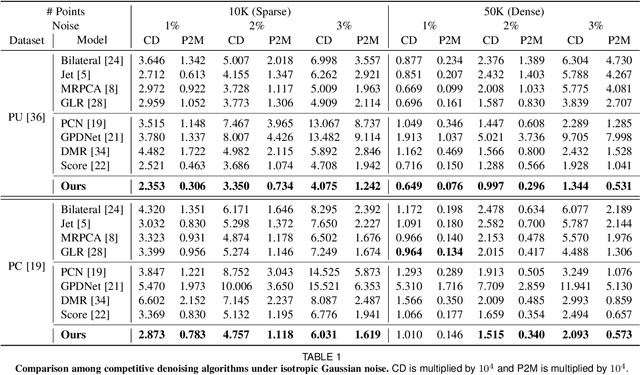
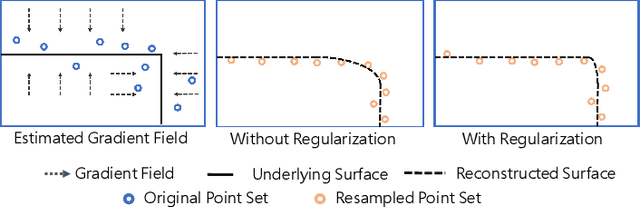
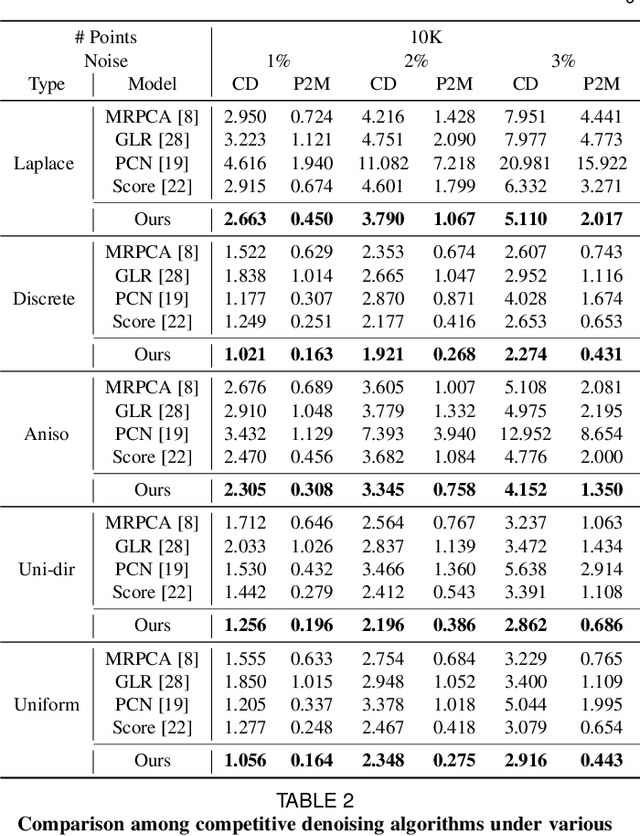
3D point clouds acquired by scanning real-world objects or scenes have found a wide range of applications including immersive telepresence, autonomous driving, surveillance, etc. They are often perturbed by noise or suffer from low density, which obstructs downstream tasks such as surface reconstruction and understanding. In this paper, we propose a novel paradigm of point set resampling for restoration, which learns continuous gradient fields of point clouds that converge points towards the underlying surface. In particular, we represent a point cloud via its gradient field -- the gradient of the log-probability density function, and enforce the gradient field to be continuous, thus guaranteeing the continuity of the model for solvable optimization. Based on the continuous gradient fields estimated via a proposed neural network, resampling a point cloud amounts to performing gradient-based Markov Chain Monte Carlo (MCMC) on the input noisy or sparse point cloud. Further, we propose to introduce regularization into the gradient-based MCMC during point cloud restoration, which essentially refines the intermediate resampled point cloud iteratively and accommodates various priors in the resampling process. Extensive experimental results demonstrate that the proposed point set resampling achieves the state-of-the-art performance in representative restoration tasks including point cloud denoising and upsampling.
Score-Based Point Cloud Denoising
Aug 15, 2021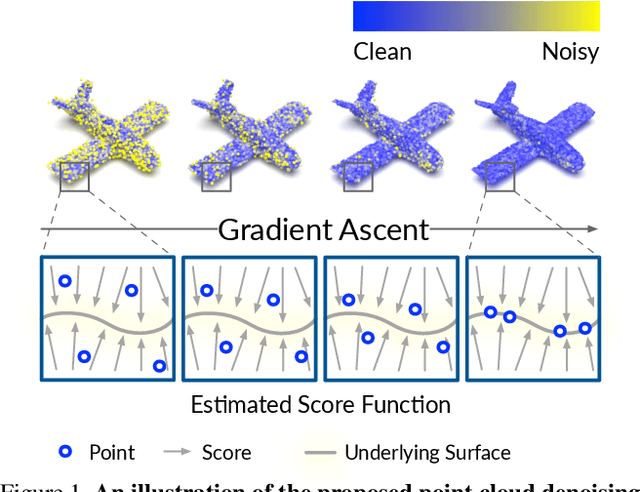
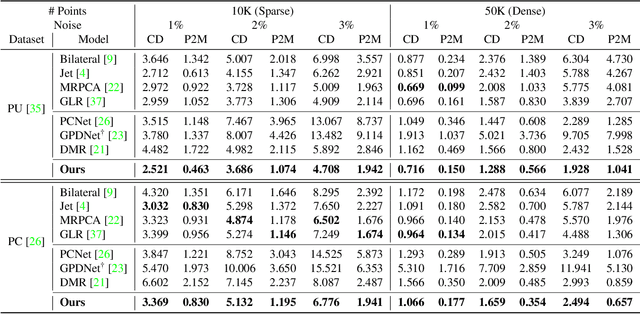


Point clouds acquired from scanning devices are often perturbed by noise, which affects downstream tasks such as surface reconstruction and analysis. The distribution of a noisy point cloud can be viewed as the distribution of a set of noise-free samples $p(x)$ convolved with some noise model $n$, leading to $(p * n)(x)$ whose mode is the underlying clean surface. To denoise a noisy point cloud, we propose to increase the log-likelihood of each point from $p * n$ via gradient ascent -- iteratively updating each point's position. Since $p * n$ is unknown at test-time, and we only need the score (i.e., the gradient of the log-probability function) to perform gradient ascent, we propose a neural network architecture to estimate the score of $p * n$ given only noisy point clouds as input. We derive objective functions for training the network and develop a denoising algorithm leveraging on the estimated scores. Experiments demonstrate that the proposed model outperforms state-of-the-art methods under a variety of noise models, and shows the potential to be applied in other tasks such as point cloud upsampling. The code is available at \url{https://github.com/luost26/score-denoise}.
Learning Gradient Fields for Molecular Conformation Generation
Jun 07, 2021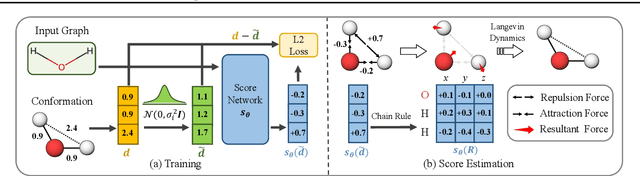
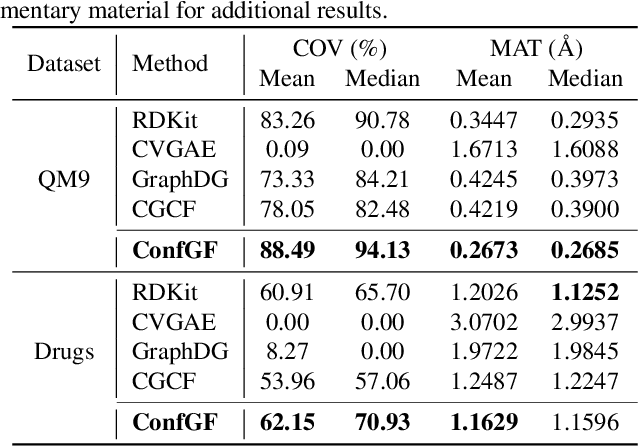
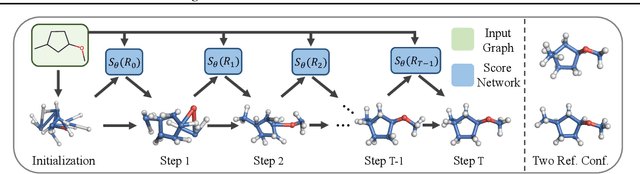
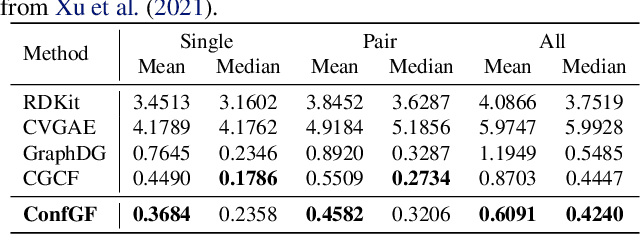
We study a fundamental problem in computational chemistry known as molecular conformation generation, trying to predict stable 3D structures from 2D molecular graphs. Existing machine learning approaches usually first predict distances between atoms and then generate a 3D structure satisfying the distances, where noise in predicted distances may induce extra errors during 3D coordinate generation. Inspired by the traditional force field methods for molecular dynamics simulation, in this paper, we propose a novel approach called ConfGF by directly estimating the gradient fields of the log density of atomic coordinates. The estimated gradient fields allow directly generating stable conformations via Langevin dynamics. However, the problem is very challenging as the gradient fields are roto-translation equivariant. We notice that estimating the gradient fields of atomic coordinates can be translated to estimating the gradient fields of interatomic distances, and hence develop a novel algorithm based on recent score-based generative models to effectively estimate these gradients. Experimental results across multiple tasks show that ConfGF outperforms previous state-of-the-art baselines by a significant margin.