 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAtomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
Mar 18, 2026Understanding the dynamic behavior of biomolecules is fundamental to elucidating biological function and facilitating drug discovery. While Molecular Dynamics (MD) simulations provide a rigorous physical basis for studying these dynamics, they remain computationally expensive for long timescales. Conversely, recent deep generative models accelerate conformation generation but are typically either failing to model temporal relationship or built only for monomeric proteins. To bridge this gap, we introduce ATMOS, a novel generative framework based on State Space Models (SSM) designed to generate atom-level MD trajectories for biomolecular systems. ATMOS integrates a Pairformer-based state transition mechanism to capture long-range temporal dependencies, with a diffusion-based module to decode trajectory frames in an autoregressive manner. ATMOS is trained across crystal structures from PDB and conformation trajectory from large-scale MD simulation datasets including mdCATH and MISATO. We demonstrate that ATMOS achieves state-of-the-art performance in generating conformation trajectories for both protein monomers and complex protein-ligand systems. By enabling efficient inference of atomic trajectory of motions, this work establishes a promising foundation for modeling biomolecular dynamics.
Structure Language Models for Protein Conformation Generation
Oct 24, 2024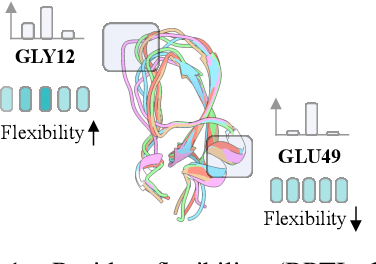
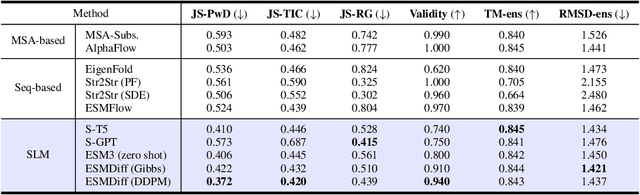
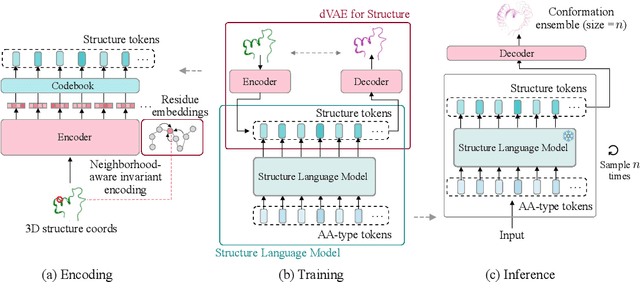
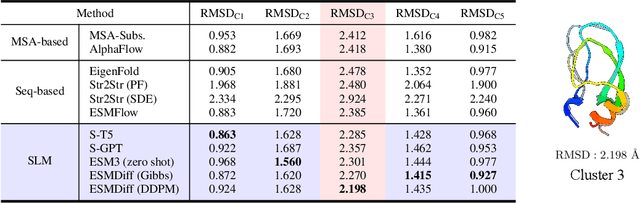
Proteins adopt multiple structural conformations to perform their diverse biological functions, and understanding these conformations is crucial for advancing drug discovery. Traditional physics-based simulation methods often struggle with sampling equilibrium conformations and are computationally expensive. Recently, deep generative models have shown promise in generating protein conformations as a more efficient alternative. However, these methods predominantly rely on the diffusion process within a 3D geometric space, which typically centers around the vicinity of metastable states and is often inefficient in terms of runtime. In this paper, we introduce Structure Language Modeling (SLM) as a novel framework for efficient protein conformation generation. Specifically, the protein structures are first encoded into a compact latent space using a discrete variational auto-encoder, followed by conditional language modeling that effectively captures sequence-specific conformation distributions. This enables a more efficient and interpretable exploration of diverse ensemble modes compared to existing methods. Based on this general framework, we instantiate SLM with various popular LM architectures as well as proposing the ESMDiff, a novel BERT-like structure language model fine-tuned from ESM3 with masked diffusion. We verify our approach in various scenarios, including the equilibrium dynamics of BPTI, conformational change pairs, and intrinsically disordered proteins. SLM provides a highly efficient solution, offering a 20-100x speedup than existing methods in generating diverse conformations, shedding light on promising avenues for future research.
Fusing Neural and Physical: Augment Protein Conformation Sampling with Tractable Simulations
Feb 16, 2024The protein dynamics are common and important for their biological functions and properties, the study of which usually involves time-consuming molecular dynamics (MD) simulations in silico. Recently, generative models has been leveraged as a surrogate sampler to obtain conformation ensembles with orders of magnitude faster and without requiring any simulation data (a "zero-shot" inference). However, being agnostic of the underlying energy landscape, the accuracy of such generative model may still be limited. In this work, we explore the few-shot setting of such pre-trained generative sampler which incorporates MD simulations in a tractable manner. Specifically, given a target protein of interest, we first acquire some seeding conformations from the pre-trained sampler followed by a number of physical simulations in parallel starting from these seeding samples. Then we fine-tuned the generative model using the simulation trajectories above to become a target-specific sampler. Experimental results demonstrated the superior performance of such few-shot conformation sampler at a tractable computational cost.
Protein Sequence and Structure Co-Design with Equivariant Translation
Oct 17, 2022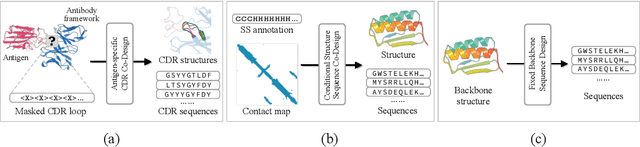
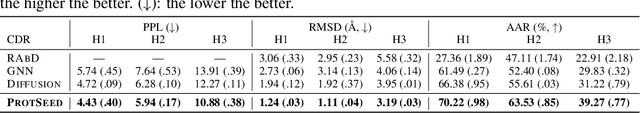
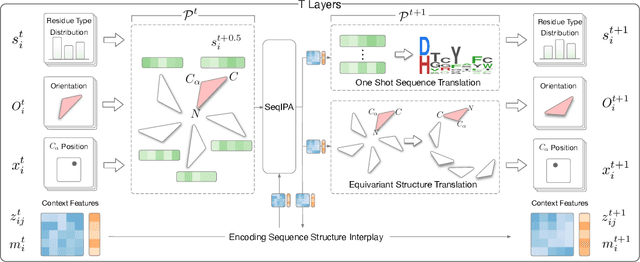
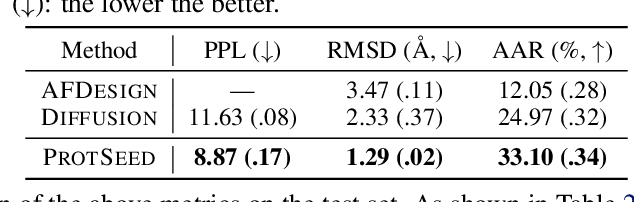
Proteins are macromolecules that perform essential functions in all living organisms. Designing novel proteins with specific structures and desired functions has been a long-standing challenge in the field of bioengineering. Existing approaches generate both protein sequence and structure using either autoregressive models or diffusion models, both of which suffer from high inference costs. In this paper, we propose a new approach capable of protein sequence and structure co-design, which iteratively translates both protein sequence and structure into the desired state from random initialization, based on context features given a priori. Our model consists of a trigonometry-aware encoder that reasons geometrical constraints and interactions from context features, and a roto-translation equivariant decoder that translates protein sequence and structure interdependently. Notably, all protein amino acids are updated in one shot in each translation step, which significantly accelerates the inference process. Experimental results across multiple tasks show that our model outperforms previous state-of-the-art baselines by a large margin, and is able to design proteins of high fidelity as regards both sequence and structure, with running time orders of magnitude less than sampling-based methods.
E3Bind: An End-to-End Equivariant Network for Protein-Ligand Docking
Oct 12, 2022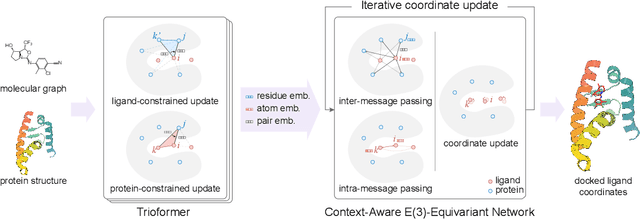
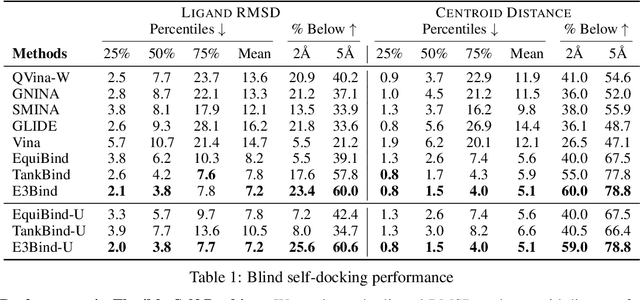
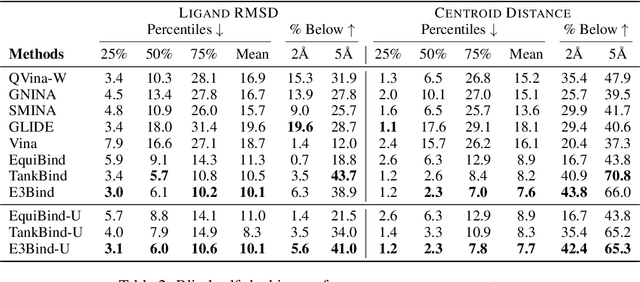
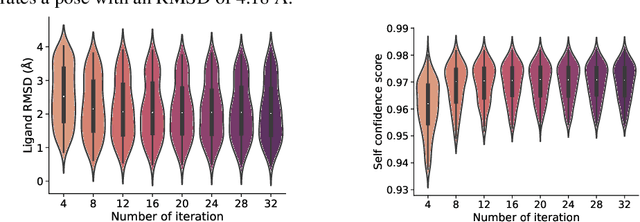
In silico prediction of the ligand binding pose to a given protein target is a crucial but challenging task in drug discovery. This work focuses on blind flexible selfdocking, where we aim to predict the positions, orientations and conformations of docked molecules. Traditional physics-based methods usually suffer from inaccurate scoring functions and high inference cost. Recently, data-driven methods based on deep learning techniques are attracting growing interest thanks to their efficiency during inference and promising performance. These methods usually either adopt a two-stage approach by first predicting the distances between proteins and ligands and then generating the final coordinates based on the predicted distances, or directly predicting the global roto-translation of ligands. In this paper, we take a different route. Inspired by the resounding success of AlphaFold2 for protein structure prediction, we propose E3Bind, an end-to-end equivariant network that iteratively updates the ligand pose. E3Bind models the protein-ligand interaction through careful consideration of the geometric constraints in docking and the local context of the binding site. Experiments on standard benchmark datasets demonstrate the superior performance of our end-to-end trainable model compared to traditional and recently-proposed deep learning methods.
A Roadmap for Big Model
Apr 02, 2022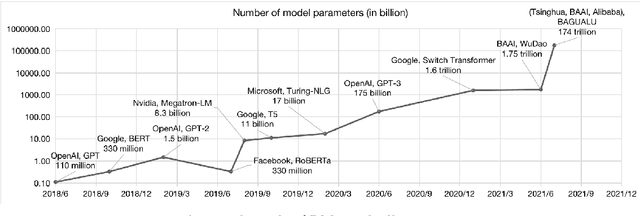
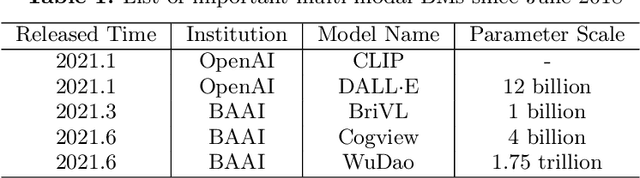
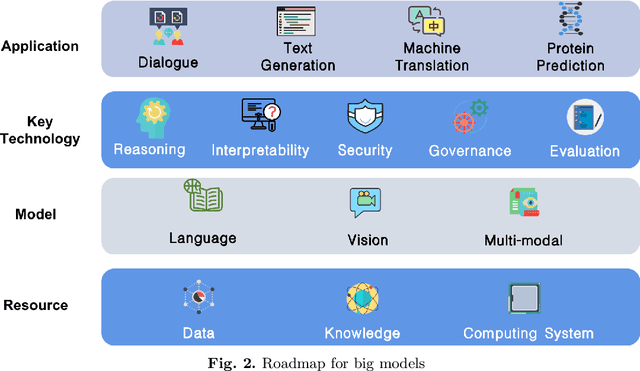
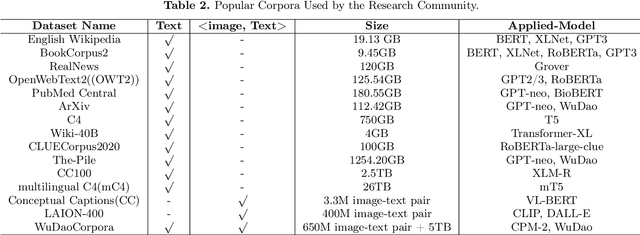
With the rapid development of deep learning, training Big Models (BMs) for multiple downstream tasks becomes a popular paradigm. Researchers have achieved various outcomes in the construction of BMs and the BM application in many fields. At present, there is a lack of research work that sorts out the overall progress of BMs and guides the follow-up research. In this paper, we cover not only the BM technologies themselves but also the prerequisites for BM training and applications with BMs, dividing the BM review into four parts: Resource, Models, Key Technologies and Application. We introduce 16 specific BM-related topics in those four parts, they are Data, Knowledge, Computing System, Parallel Training System, Language Model, Vision Model, Multi-modal Model, Theory&Interpretability, Commonsense Reasoning, Reliability&Security, Governance, Evaluation, Machine Translation, Text Generation, Dialogue and Protein Research. In each topic, we summarize clearly the current studies and propose some future research directions. At the end of this paper, we conclude the further development of BMs in a more general view.
GeoDiff: a Geometric Diffusion Model for Molecular Conformation Generation
Mar 06, 2022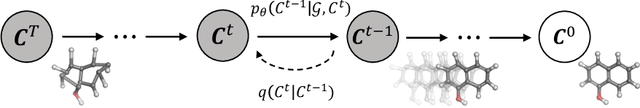
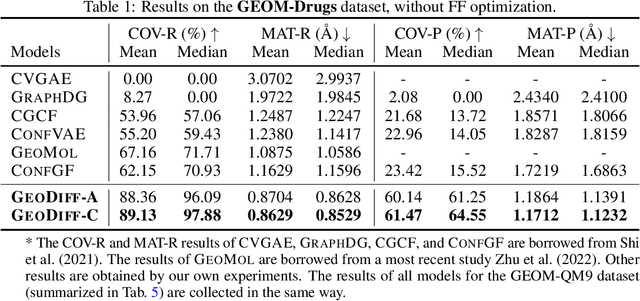
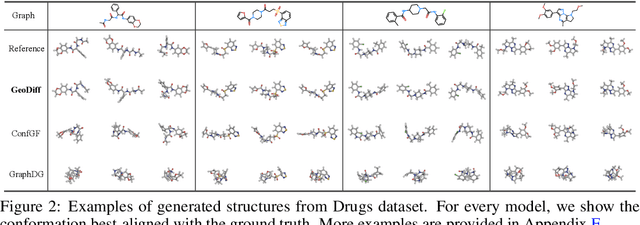

Predicting molecular conformations from molecular graphs is a fundamental problem in cheminformatics and drug discovery. Recently, significant progress has been achieved with machine learning approaches, especially with deep generative models. Inspired by the diffusion process in classical non-equilibrium thermodynamics where heated particles will diffuse from original states to a noise distribution, in this paper, we propose a novel generative model named GeoDiff for molecular conformation prediction. GeoDiff treats each atom as a particle and learns to directly reverse the diffusion process (i.e., transforming from a noise distribution to stable conformations) as a Markov chain. Modeling such a generation process is however very challenging as the likelihood of conformations should be roto-translational invariant. We theoretically show that Markov chains evolving with equivariant Markov kernels can induce an invariant distribution by design, and further propose building blocks for the Markov kernels to preserve the desirable equivariance property. The whole framework can be efficiently trained in an end-to-end fashion by optimizing a weighted variational lower bound to the (conditional) likelihood. Experiments on multiple benchmarks show that GeoDiff is superior or comparable to existing state-of-the-art approaches, especially on large molecules.
TorchDrug: A Powerful and Flexible Machine Learning Platform for Drug Discovery
Feb 16, 2022

Machine learning has huge potential to revolutionize the field of drug discovery and is attracting increasing attention in recent years. However, lacking domain knowledge (e.g., which tasks to work on), standard benchmarks and data preprocessing pipelines are the main obstacles for machine learning researchers to work in this domain. To facilitate the progress of machine learning for drug discovery, we develop TorchDrug, a powerful and flexible machine learning platform for drug discovery built on top of PyTorch. TorchDrug benchmarks a variety of important tasks in drug discovery, including molecular property prediction, pretrained molecular representations, de novo molecular design and optimization, retrosynthsis prediction, and biomedical knowledge graph reasoning. State-of-the-art techniques based on geometric deep learning (or graph machine learning), deep generative models, reinforcement learning and knowledge graph reasoning are implemented for these tasks. TorchDrug features a hierarchical interface that facilitates customization from both novices and experts in this domain. Tutorials, benchmark results and documentation are available at https://torchdrug.ai. Code is released under Apache License 2.0.
Non-Autoregressive Electron Redistribution Modeling for Reaction Prediction
Jun 08, 2021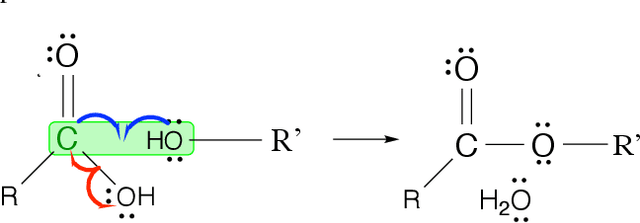
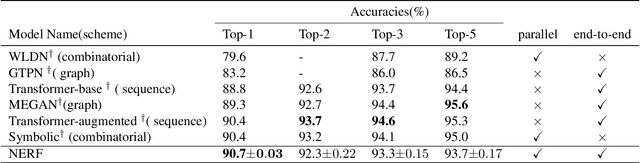
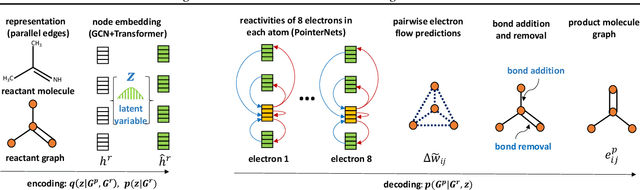
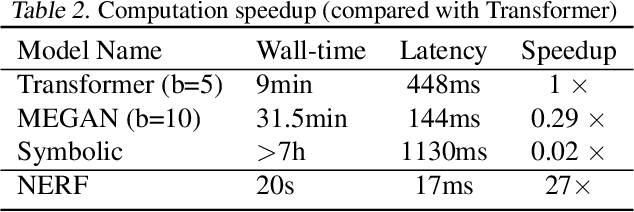
Reliably predicting the products of chemical reactions presents a fundamental challenge in synthetic chemistry. Existing machine learning approaches typically produce a reaction product by sequentially forming its subparts or intermediate molecules. Such autoregressive methods, however, not only require a pre-defined order for the incremental construction but preclude the use of parallel decoding for efficient computation. To address these issues, we devise a non-autoregressive learning paradigm that predicts reaction in one shot. Leveraging the fact that chemical reactions can be described as a redistribution of electrons in molecules, we formulate a reaction as an arbitrary electron flow and predict it with a novel multi-pointer decoding network. Experiments on the USPTO-MIT dataset show that our approach has established a new state-of-the-art top-1 accuracy and achieves at least 27 times inference speedup over the state-of-the-art methods. Also, our predictions are easier for chemists to interpret owing to predicting the electron flows.
Learning Gradient Fields for Molecular Conformation Generation
Jun 07, 2021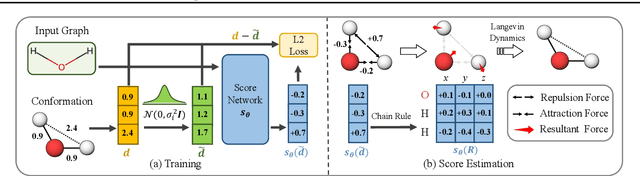
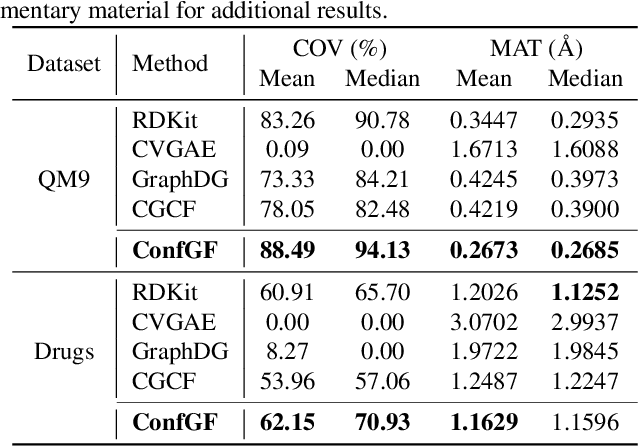
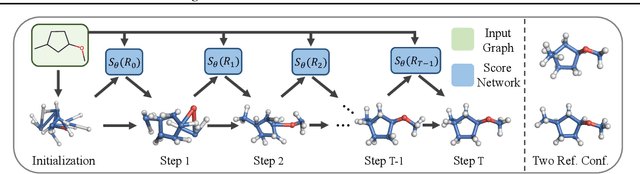
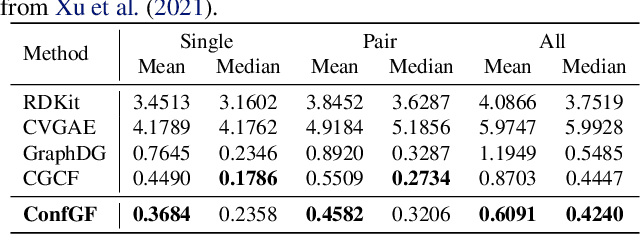
We study a fundamental problem in computational chemistry known as molecular conformation generation, trying to predict stable 3D structures from 2D molecular graphs. Existing machine learning approaches usually first predict distances between atoms and then generate a 3D structure satisfying the distances, where noise in predicted distances may induce extra errors during 3D coordinate generation. Inspired by the traditional force field methods for molecular dynamics simulation, in this paper, we propose a novel approach called ConfGF by directly estimating the gradient fields of the log density of atomic coordinates. The estimated gradient fields allow directly generating stable conformations via Langevin dynamics. However, the problem is very challenging as the gradient fields are roto-translation equivariant. We notice that estimating the gradient fields of atomic coordinates can be translated to estimating the gradient fields of interatomic distances, and hence develop a novel algorithm based on recent score-based generative models to effectively estimate these gradients. Experimental results across multiple tasks show that ConfGF outperforms previous state-of-the-art baselines by a significant margin.