 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEffective Protein-Protein Interaction Exploration with PPIretrieval
Feb 06, 2024Protein-protein interactions (PPIs) are crucial in regulating numerous cellular functions, including signal transduction, transportation, and immune defense. As the accuracy of multi-chain protein complex structure prediction improves, the challenge has shifted towards effectively navigating the vast complex universe to identify potential PPIs. Herein, we propose PPIretrieval, the first deep learning-based model for protein-protein interaction exploration, which leverages existing PPI data to effectively search for potential PPIs in an embedding space, capturing rich geometric and chemical information of protein surfaces. When provided with an unseen query protein with its associated binding site, PPIretrieval effectively identifies a potential binding partner along with its corresponding binding site in an embedding space, facilitating the formation of protein-protein complexes.
Non-Autoregressive Electron Redistribution Modeling for Reaction Prediction
Jun 08, 2021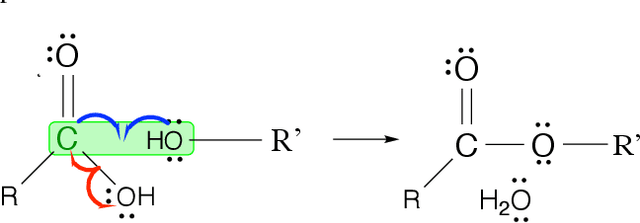
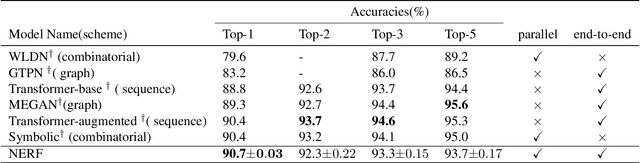
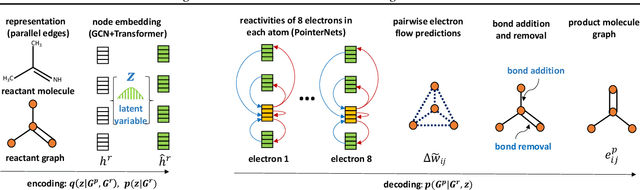
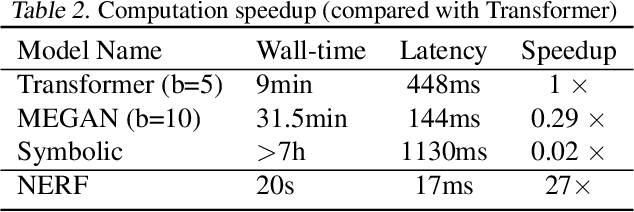
Reliably predicting the products of chemical reactions presents a fundamental challenge in synthetic chemistry. Existing machine learning approaches typically produce a reaction product by sequentially forming its subparts or intermediate molecules. Such autoregressive methods, however, not only require a pre-defined order for the incremental construction but preclude the use of parallel decoding for efficient computation. To address these issues, we devise a non-autoregressive learning paradigm that predicts reaction in one shot. Leveraging the fact that chemical reactions can be described as a redistribution of electrons in molecules, we formulate a reaction as an arbitrary electron flow and predict it with a novel multi-pointer decoding network. Experiments on the USPTO-MIT dataset show that our approach has established a new state-of-the-art top-1 accuracy and achieves at least 27 times inference speedup over the state-of-the-art methods. Also, our predictions are easier for chemists to interpret owing to predicting the electron flows.
Are Learned Molecular Representations Ready For Prime Time?
Apr 02, 2019



Advancements in neural machinery have led to a wide range of algorithmic solutions for molecular property prediction. Two classes of models in particular have yielded promising results: neural networks applied to computed molecular fingerprints or expert-crafted descriptors, and graph convolutional neural networks that construct a learned molecular representation by operating on the graph structure of the molecule. However, recent literature has yet to clearly determine which of these two methods is superior when generalizing to new chemical space. Furthermore, prior research has rarely examined these new models in industry research settings in comparison to existing employed models. In this paper, we benchmark models extensively on 19 public and 15 proprietary industrial datasets spanning a wide variety of chemical endpoints. In addition, we introduce a graph convolutional model that consistently outperforms models using fixed molecular descriptors as well as previous graph neural architectures on both public and proprietary datasets. Our empirical findings indicate that while approaches based on these representations have yet to reach the level of experimental reproducibility, our proposed model nevertheless offers significant improvements over models currently used in industrial workflows.