 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting Immune Biomarkers with MultiModal Mixture-of-Expert Pathology Foundation Models Empowers Precision Oncology
Jun 16, 2026Predicting immune biomarkers associated with the tumor immune microenvironment (TIME) is critical for advancing precision oncology, yet existing approaches are largely limited to single image modalities and suffer from insufficient resolution and incomplete utilization of complementary clinical and biological information. Here we introduce MixTIME, a multimodal foundation model that leverages a mixture-of-experts (MoE) architecture to integrate pathology foundation models trained across distinct modalities: image only (UNIv2), image text (CONCHv1.5), and image transcriptomic (STPath) representations for pixel-level and slide-level prediction of multiplex immunofluorescence (mIF) protein expression from hematoxylin and eosin (HE) whole-slide images. MixTIME employs a learnable router to dynamically weight expert contributions and is trained with a distribution- and tendency-aware loss function. Benchmarked on two datasets of different scales, MixTIME achieves state-of-the-art performance across 17 protein markers as measured by correlation metrics. The predicted mIF profiles substantially enhance downstream tasks, including spatial domain identification, survival prediction, and AI-assisted pathology report generation validated by expert pathologists from multiple institutes across the world. Furthermore, MixTIME enables longitudinal tracking of protein expression dynamics across clinical time points and reveals protein gene interaction patterns linked to drug resistance and immune suppression in tumor microenvironments. Collectively, MixTIME provides a scalable framework for multimodal biomarker discovery and clinical translation in computational pathology.
Benchmarking AI Agents for Addressing Scientific Challenges Across Scales
Jun 10, 2026AI agents are increasingly being developed to accelerate scientific discovery, yet their practical capabilities in real research settings remain poorly understood. Existing benchmarks for AI agents rarely capture the complexity, heterogeneity, and extended reasoning required by scientific work, whereas benchmarks for scientific tasks often reduce research to static, direct problems and provide limited support for interactive evaluation. Here, we introduce SciAgentArena, a systematic benchmark for evaluating AI agents in real-world scientific research scenarios drawn from emerging needs across multiple domains. SciAgentArena comprises approximately 200 tasks with stepwise verification and an interactive, agent-agnostic environment for assessing diverse AI agents. Using this benchmark, we find that current agents can contribute effectively to well-specified data-analysis workflows, particularly when the task structure and evaluation criteria are clear. However, their performance remains uneven across scientific contexts: agents struggle to generate genuinely novel insights, sustain self-directed exploration, and formulate robust solutions for open-ended research questions. We further characterize common failure modes across agents and identify opportunities for improving their reliability, autonomy, and scientific reasoning. Together, SciAgentArena provides a practical framework for measuring progress in AI agents for science and for guiding the design of future agents capable of addressing complex scientific challenges. Full codes, tasks, and datasets can be accessed via this link: https://sciagentarena.github.io/.
BindEnergyCraft: Casting Protein Structure Predictors as Energy-Based Models for Binder Design
May 27, 2025Protein binder design has been transformed by hallucination-based methods that optimize structure prediction confidence metrics, such as the interface predicted TM-score (ipTM), via backpropagation. However, these metrics do not reflect the statistical likelihood of a binder-target complex under the learned distribution and yield sparse gradients for optimization. In this work, we propose a method to extract such likelihoods from structure predictors by reinterpreting their confidence outputs as an energy-based model (EBM). By leveraging the Joint Energy-based Modeling (JEM) framework, we introduce pTMEnergy, a statistical energy function derived from predicted inter-residue error distributions. We incorporate pTMEnergy into BindEnergyCraft (BECraft), a design pipeline that maintains the same optimization framework as BindCraft but replaces ipTM with our energy-based objective. BECraft outperforms BindCraft, RFDiffusion, and ESM3 across multiple challenging targets, achieving higher in silico binder success rates while reducing structural clashes. Furthermore, pTMEnergy establishes a new state-of-the-art in structure-based virtual screening tasks for miniprotein and RNA aptamer binders.
Reaction-conditioned De Novo Enzyme Design with GENzyme
Nov 10, 2024The introduction of models like RFDiffusionAA, AlphaFold3, AlphaProteo, and Chai1 has revolutionized protein structure modeling and interaction prediction, primarily from a binding perspective, focusing on creating ideal lock-and-key models. However, these methods can fall short for enzyme-substrate interactions, where perfect binding models are rare, and induced fit states are more common. To address this, we shift to a functional perspective for enzyme design, where the enzyme function is defined by the reaction it catalyzes. Here, we introduce \textsc{GENzyme}, a \textit{de novo} enzyme design model that takes a catalytic reaction as input and generates the catalytic pocket, full enzyme structure, and enzyme-substrate binding complex. \textsc{GENzyme} is an end-to-end, three-staged model that integrates (1) a catalytic pocket generation and sequence co-design module, (2) a pocket inpainting and enzyme inverse folding module, and (3) a binding and screening module to optimize and predict enzyme-substrate complexes. The entire design process is driven by the catalytic reaction being targeted. This reaction-first approach allows for more accurate and biologically relevant enzyme design, potentially surpassing structure-based and binding-focused models in creating enzymes capable of catalyzing specific reactions. We provide \textsc{GENzyme} code at https://github.com/WillHua127/GENzyme.
Boltzmann-Aligned Inverse Folding Model as a Predictor of Mutational Effects on Protein-Protein Interactions
Oct 12, 2024



Predicting the change in binding free energy ($\Delta \Delta G$) is crucial for understanding and modulating protein-protein interactions, which are critical in drug design. Due to the scarcity of experimental $\Delta \Delta G$ data, existing methods focus on pre-training, while neglecting the importance of alignment. In this work, we propose the Boltzmann Alignment technique to transfer knowledge from pre-trained inverse folding models to $\Delta \Delta G$ prediction. We begin by analyzing the thermodynamic definition of $\Delta \Delta G$ and introducing the Boltzmann distribution to connect energy with protein conformational distribution. However, the protein conformational distribution is intractable; therefore, we employ Bayes' theorem to circumvent direct estimation and instead utilize the log-likelihood provided by protein inverse folding models for $\Delta \Delta G$ estimation. Compared to previous inverse folding-based methods, our method explicitly accounts for the unbound state of protein complex in the $\Delta \Delta G$ thermodynamic cycle, introducing a physical inductive bias and achieving both supervised and unsupervised state-of-the-art (SoTA) performance. Experimental results on SKEMPI v2 indicate that our method achieves Spearman coefficients of 0.3201 (unsupervised) and 0.5134 (supervised), significantly surpassing the previously reported SoTA values of 0.2632 and 0.4324, respectively. Futhermore, we demonstrate the capability of our method on binding energy prediction, protein-protein docking and antibody optimization tasks.
RNAFlow: RNA Structure & Sequence Design via Inverse Folding-Based Flow Matching
May 29, 2024The growing significance of RNA engineering in diverse biological applications has spurred interest in developing AI methods for structure-based RNA design. While diffusion models have excelled in protein design, adapting them for RNA presents new challenges due to RNA's conformational flexibility and the computational cost of fine-tuning large structure prediction models. To this end, we propose RNAFlow, a flow matching model for protein-conditioned RNA sequence-structure design. Its denoising network integrates an RNA inverse folding model and a pre-trained RosettaFold2NA network for generation of RNA sequences and structures. The integration of inverse folding in the structure denoising process allows us to simplify training by fixing the structure prediction network. We further enhance the inverse folding model by conditioning it on inferred conformational ensembles to model dynamic RNA conformations. Evaluation on protein-conditioned RNA structure and sequence generation tasks demonstrates RNAFlow's advantage over existing RNA design methods.
Generative Enzyme Design Guided by Functionally Important Sites and Small-Molecule Substrates
May 13, 2024Enzymes are genetically encoded biocatalysts capable of accelerating chemical reactions. How can we automatically design functional enzymes? In this paper, we propose EnzyGen, an approach to learn a unified model to design enzymes across all functional families. Our key idea is to generate an enzyme's amino acid sequence and their three-dimensional (3D) coordinates based on functionally important sites and substrates corresponding to a desired catalytic function. These sites are automatically mined from enzyme databases. EnzyGen consists of a novel interleaving network of attention and neighborhood equivariant layers, which captures both long-range correlation in an entire protein sequence and local influence from nearest amino acids in 3D space. To learn the generative model, we devise a joint training objective, including a sequence generation loss, a position prediction loss and an enzyme-substrate interaction loss. We further construct EnzyBench, a dataset with 3157 enzyme families, covering all available enzymes within the protein data bank (PDB). Experimental results show that our EnzyGen consistently achieves the best performance across all 323 testing families, surpassing the best baseline by 10.79% in terms of substrate binding affinity. These findings demonstrate EnzyGen's superior capability in designing well-folded and effective enzymes binding to specific substrates with high affinities.
SurfPro: Functional Protein Design Based on Continuous Surface
May 07, 2024How can we design proteins with desired functions? We are motivated by a chemical intuition that both geometric structure and biochemical properties are critical to a protein's function. In this paper, we propose SurfPro, a new method to generate functional proteins given a desired surface and its associated biochemical properties. SurfPro comprises a hierarchical encoder that progressively models the geometric shape and biochemical features of a protein surface, and an autoregressive decoder to produce an amino acid sequence. We evaluate SurfPro on a standard inverse folding benchmark CATH 4.2 and two functional protein design tasks: protein binder design and enzyme design. Our SurfPro consistently surpasses previous state-of-the-art inverse folding methods, achieving a recovery rate of 57.78% on CATH 4.2 and higher success rates in terms of protein-protein binding and enzyme-substrate interaction scores.
Unsupervised Protein-Ligand Binding Energy Prediction via Neural Euler's Rotation Equation
Jan 25, 2023



Protein-ligand binding prediction is a fundamental problem in AI-driven drug discovery. Prior work focused on supervised learning methods using a large set of binding affinity data for small molecules, but it is hard to apply the same strategy to other drug classes like antibodies as labelled data is limited. In this paper, we explore unsupervised approaches and reformulate binding energy prediction as a generative modeling task. Specifically, we train an energy-based model on a set of unlabelled protein-ligand complexes using SE(3) denoising score matching and interpret its log-likelihood as binding affinity. Our key contribution is a new equivariant rotation prediction network called Neural Euler's Rotation Equations (NERE) for SE(3) score matching. It predicts a rotation by modeling the force and torque between protein and ligand atoms, where the force is defined as the gradient of an energy function with respect to atom coordinates. We evaluate NERE on protein-ligand and antibody-antigen binding affinity prediction benchmarks. Our model outperforms all unsupervised baselines (physics-based and statistical potentials) and matches supervised learning methods in the antibody case.
Antibody-Antigen Docking and Design via Hierarchical Equivariant Refinement
Jul 14, 2022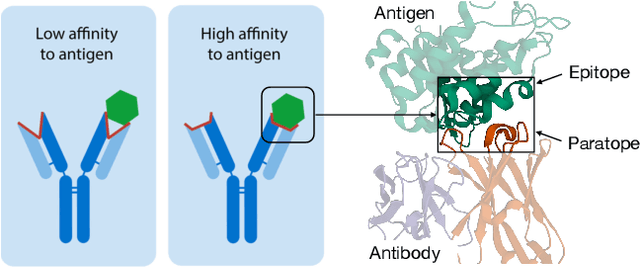
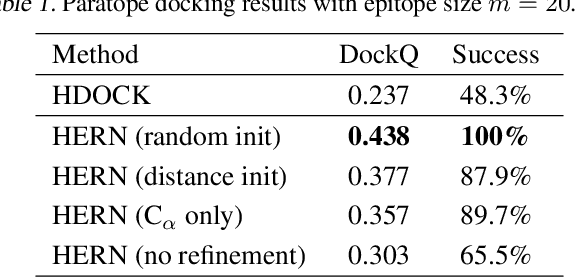
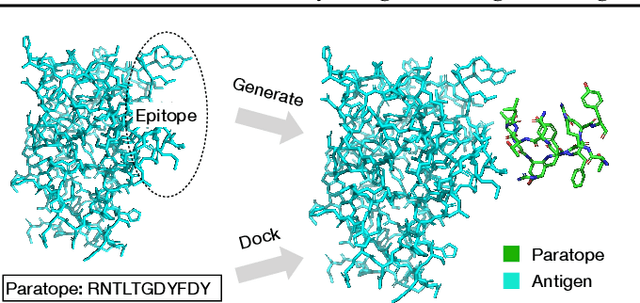
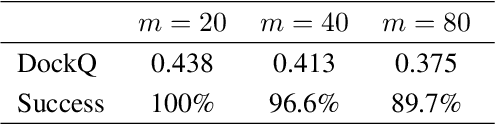
Computational antibody design seeks to automatically create an antibody that binds to an antigen. The binding affinity is governed by the 3D binding interface where antibody residues (paratope) closely interact with antigen residues (epitope). Thus, predicting 3D paratope-epitope complex (docking) is the key to finding the best paratope. In this paper, we propose a new model called Hierarchical Equivariant Refinement Network (HERN) for paratope docking and design. During docking, HERN employs a hierarchical message passing network to predict atomic forces and use them to refine a binding complex in an iterative, equivariant manner. During generation, its autoregressive decoder progressively docks generated paratopes and builds a geometric representation of the binding interface to guide the next residue choice. Our results show that HERN significantly outperforms prior state-of-the-art on paratope docking and design benchmarks.