 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInternAgent-1.5: A Unified Agentic Framework for Long-Horizon Autonomous Scientific Discovery
Feb 09, 2026We introduce InternAgent-1.5, a unified system designed for end-to-end scientific discovery across computational and empirical domains. The system is built on a structured architecture composed of three coordinated subsystems for generation, verification, and evolution. These subsystems are supported by foundational capabilities for deep research, solution optimization, and long horizon memory. The architecture allows InternAgent-1.5 to operate continuously across extended discovery cycles while maintaining coherent and improving behavior. It also enables the system to coordinate computational modeling and laboratory experimentation within a single unified system. We evaluate InternAgent-1.5 on scientific reasoning benchmarks such as GAIA, HLE, GPQA, and FrontierScience, and the system achieves leading performance that demonstrates strong foundational capabilities. Beyond these benchmarks, we further assess two categories of discovery tasks. In algorithm discovery tasks, InternAgent-1.5 autonomously designs competitive methods for core machine learning problems. In empirical discovery tasks, it executes complete computational or wet lab experiments and produces scientific findings in earth, life, biological, and physical domains. Overall, these results show that InternAgent-1.5 provides a general and scalable framework for autonomous scientific discovery.
Efficient Protein Optimization via Structure-aware Hamiltonian Dynamics
Jan 16, 2026The ability to engineer optimized protein variants has transformative potential for biotechnology and medicine. Prior sequence-based optimization methods struggle with the high-dimensional complexities due to the epistasis effect and the disregard for structural constraints. To address this, we propose HADES, a Bayesian optimization method utilizing Hamiltonian dynamics to efficiently sample from a structure-aware approximated posterior. Leveraging momentum and uncertainty in the simulated physical movements, HADES enables rapid transition of proposals toward promising areas. A position discretization procedure is introduced to propose discrete protein sequences from such a continuous state system. The posterior surrogate is powered by a two-stage encoder-decoder framework to determine the structure and function relationships between mutant neighbors, consequently learning a smoothed landscape to sample from. Extensive experiments demonstrate that our method outperforms state-of-the-art baselines in in-silico evaluations across most metrics. Remarkably, our approach offers a unique advantage by leveraging the mutual constraints between protein structure and sequence, facilitating the design of protein sequences with similar structures and optimized properties. The code and data are publicly available at https://github.com/GENTEL-lab/HADES.
EnerBridge-DPO: Energy-Guided Protein Inverse Folding with Markov Bridges and Direct Preference Optimization
Jun 11, 2025


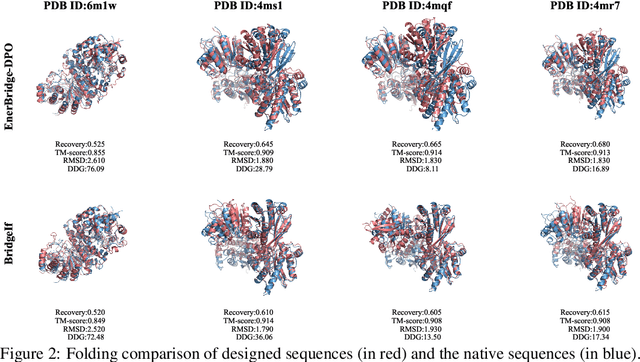
Designing protein sequences with optimal energetic stability is a key challenge in protein inverse folding, as current deep learning methods are primarily trained by maximizing sequence recovery rates, often neglecting the energy of the generated sequences. This work aims to overcome this limitation by developing a model that directly generates low-energy, stable protein sequences. We propose EnerBridge-DPO, a novel inverse folding framework focused on generating low-energy, high-stability protein sequences. Our core innovation lies in: First, integrating Markov Bridges with Direct Preference Optimization (DPO), where energy-based preferences are used to fine-tune the Markov Bridge model. The Markov Bridge initiates optimization from an information-rich prior sequence, providing DPO with a pool of structurally plausible sequence candidates. Second, an explicit energy constraint loss is introduced, which enhances the energy-driven nature of DPO based on prior sequences, enabling the model to effectively learn energy representations from a wealth of prior knowledge and directly predict sequence energy values, thereby capturing quantitative features of the energy landscape. Our evaluations demonstrate that EnerBridge-DPO can design protein complex sequences with lower energy while maintaining sequence recovery rates comparable to state-of-the-art models, and accurately predicts $\Delta \Delta G$ values between various sequences.
A 3D pocket-aware and affinity-guided diffusion model for lead optimization
Apr 29, 2025Molecular optimization, aimed at improving binding affinity or other molecular properties, is a crucial task in drug discovery that often relies on the expertise of medicinal chemists. Recently, deep learning-based 3D generative models showed promise in enhancing the efficiency of molecular optimization. However, these models often struggle to adequately consider binding affinities with protein targets during lead optimization. Herein, we propose a 3D pocket-aware and affinity-guided diffusion model, named Diffleop, to optimize molecules with enhanced binding affinity. The model explicitly incorporates the knowledge of protein-ligand binding affinity to guide the denoising sampling for molecule generation with high affinity. The comprehensive evaluations indicated that Diffleop outperforms baseline models across multiple metrics, especially in terms of binding affinity.
Reaction-conditioned De Novo Enzyme Design with GENzyme
Nov 10, 2024The introduction of models like RFDiffusionAA, AlphaFold3, AlphaProteo, and Chai1 has revolutionized protein structure modeling and interaction prediction, primarily from a binding perspective, focusing on creating ideal lock-and-key models. However, these methods can fall short for enzyme-substrate interactions, where perfect binding models are rare, and induced fit states are more common. To address this, we shift to a functional perspective for enzyme design, where the enzyme function is defined by the reaction it catalyzes. Here, we introduce \textsc{GENzyme}, a \textit{de novo} enzyme design model that takes a catalytic reaction as input and generates the catalytic pocket, full enzyme structure, and enzyme-substrate binding complex. \textsc{GENzyme} is an end-to-end, three-staged model that integrates (1) a catalytic pocket generation and sequence co-design module, (2) a pocket inpainting and enzyme inverse folding module, and (3) a binding and screening module to optimize and predict enzyme-substrate complexes. The entire design process is driven by the catalytic reaction being targeted. This reaction-first approach allows for more accurate and biologically relevant enzyme design, potentially surpassing structure-based and binding-focused models in creating enzymes capable of catalyzing specific reactions. We provide \textsc{GENzyme} code at https://github.com/WillHua127/GENzyme.
Retrieval Augmented Diffusion Model for Structure-informed Antibody Design and Optimization
Oct 19, 2024



Antibodies are essential proteins responsible for immune responses in organisms, capable of specifically recognizing antigen molecules of pathogens. Recent advances in generative models have significantly enhanced rational antibody design. However, existing methods mainly create antibodies from scratch without template constraints, leading to model optimization challenges and unnatural sequences. To address these issues, we propose a retrieval-augmented diffusion framework, termed RADAb, for efficient antibody design. Our method leverages a set of structural homologous motifs that align with query structural constraints to guide the generative model in inversely optimizing antibodies according to desired design criteria. Specifically, we introduce a structure-informed retrieval mechanism that integrates these exemplar motifs with the input backbone through a novel dual-branch denoising module, utilizing both structural and evolutionary information. Additionally, we develop a conditional diffusion model that iteratively refines the optimization process by incorporating both global context and local evolutionary conditions. Our approach is agnostic to the choice of generative models. Empirical experiments demonstrate that our method achieves state-of-the-art performance in multiple antibody inverse folding and optimization tasks, offering a new perspective on biomolecular generative models.
Reactzyme: A Benchmark for Enzyme-Reaction Prediction
Aug 24, 2024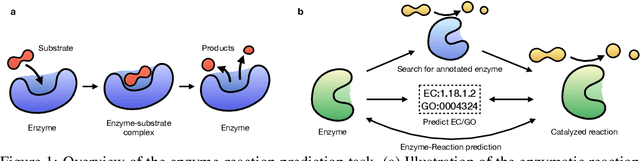
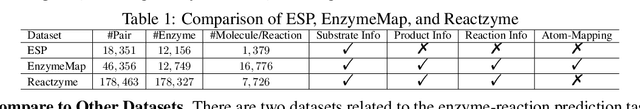
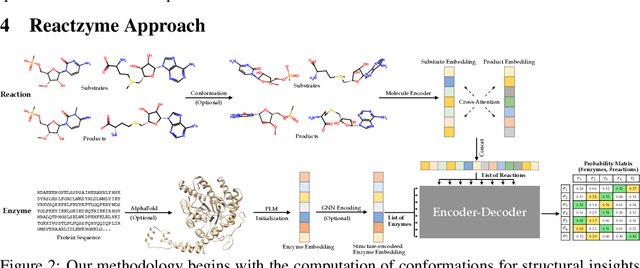
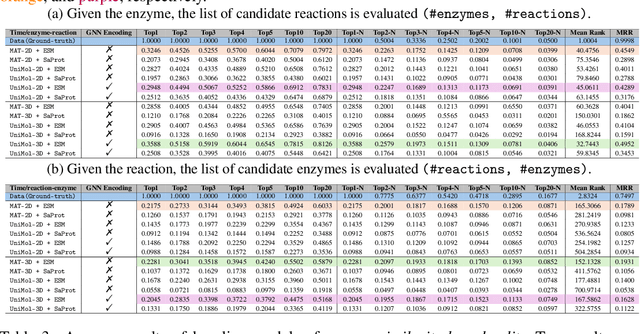
Enzymes, with their specific catalyzed reactions, are necessary for all aspects of life, enabling diverse biological processes and adaptations. Predicting enzyme functions is essential for understanding biological pathways, guiding drug development, enhancing bioproduct yields, and facilitating evolutionary studies. Addressing the inherent complexities, we introduce a new approach to annotating enzymes based on their catalyzed reactions. This method provides detailed insights into specific reactions and is adaptable to newly discovered reactions, diverging from traditional classifications by protein family or expert-derived reaction classes. We employ machine learning algorithms to analyze enzyme reaction datasets, delivering a much more refined view on the functionality of enzymes. Our evaluation leverages the largest enzyme-reaction dataset to date, derived from the SwissProt and Rhea databases with entries up to January 8, 2024. We frame the enzyme-reaction prediction as a retrieval problem, aiming to rank enzymes by their catalytic ability for specific reactions. With our model, we can recruit proteins for novel reactions and predict reactions in novel proteins, facilitating enzyme discovery and function annotation.
Incorporating Retrieval-based Causal Learning with Information Bottlenecks for Interpretable Graph Neural Networks
Feb 07, 2024



Graph Neural Networks (GNNs) have gained considerable traction for their capability to effectively process topological data, yet their interpretability remains a critical concern. Current interpretation methods are dominated by post-hoc explanations to provide a transparent and intuitive understanding of GNNs. However, they have limited performance in interpreting complicated subgraphs and can't utilize the explanation to advance GNN predictions. On the other hand, transparent GNN models are proposed to capture critical subgraphs. While such methods could improve GNN predictions, they usually don't perform well on explanations. Thus, it is desired for a new strategy to better couple GNN explanation and prediction. In this study, we have developed a novel interpretable causal GNN framework that incorporates retrieval-based causal learning with Graph Information Bottleneck (GIB) theory. The framework could semi-parametrically retrieve crucial subgraphs detected by GIB and compress the explanatory subgraphs via a causal module. The framework was demonstrated to consistently outperform state-of-the-art methods, and to achieve 32.71\% higher precision on real-world explanation scenarios with diverse explanation types. More importantly, the learned explanations were shown able to also improve GNN prediction performance.
Effective Protein-Protein Interaction Exploration with PPIretrieval
Feb 06, 2024



Protein-protein interactions (PPIs) are crucial in regulating numerous cellular functions, including signal transduction, transportation, and immune defense. As the accuracy of multi-chain protein complex structure prediction improves, the challenge has shifted towards effectively navigating the vast complex universe to identify potential PPIs. Herein, we propose PPIretrieval, the first deep learning-based model for protein-protein interaction exploration, which leverages existing PPI data to effectively search for potential PPIs in an embedding space, capturing rich geometric and chemical information of protein surfaces. When provided with an unseen query protein with its associated binding site, PPIretrieval effectively identifies a potential binding partner along with its corresponding binding site in an embedding space, facilitating the formation of protein-protein complexes.
Mixup-Augmented Meta-Learning for Sample-Efficient Fine-Tuning of Protein Simulators
Sep 07, 2023



Molecular dynamics simulations have emerged as a fundamental instrument for studying biomolecules. At the same time, it is desirable to perform simulations of a collection of particles under various conditions in which the molecules can fluctuate. In this paper, we explore and adapt the soft prompt-based learning method to molecular dynamics tasks. Our model can remarkably generalize to unseen and out-of-distribution scenarios with limited training data. While our work focuses on temperature as a test case, the versatility of our approach allows for efficient simulation through any continuous dynamic conditions, such as pressure and volumes. Our framework has two stages: 1) Pre-trains with data mixing technique, augments molecular structure data and temperature prompts, then applies a curriculum learning method by increasing the ratio of them smoothly. 2) Meta-learning-based fine-tuning framework improves sample-efficiency of fine-tuning process and gives the soft prompt-tuning better initialization points. Comprehensive experiments reveal that our framework excels in accuracy for in-domain data and demonstrates strong generalization capabilities for unseen and out-of-distribution samples.