 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePredicting sub-population specific viral evolution
Oct 28, 2024



Forecasting the change in the distribution of viral variants is crucial for therapeutic design and disease surveillance. This task poses significant modeling challenges due to the sharp differences in virus distributions across sub-populations (e.g., countries) and their dynamic interactions. Existing machine learning approaches that model the variant distribution as a whole are incapable of making location-specific predictions and ignore transmissions that shape the viral landscape. In this paper, we propose a sub-population specific protein evolution model, which predicts the time-resolved distributions of viral proteins in different locations. The algorithm explicitly models the transmission rates between sub-populations and learns their interdependence from data. The change in protein distributions across all sub-populations is defined through a linear ordinary differential equation (ODE) parametrized by transmission rates. Solving this ODE yields the likelihood of a given protein occurring in particular sub-populations. Multi-year evaluation on both SARS-CoV-2 and influenza A/H3N2 demonstrates that our model outperforms baselines in accurately predicting distributions of viral proteins across continents and countries. We also find that the transmission rates learned from data are consistent with the transmission pathways discovered by retrospective phylogenetic analysis.
Generative Enzyme Design Guided by Functionally Important Sites and Small-Molecule Substrates
May 13, 2024Enzymes are genetically encoded biocatalysts capable of accelerating chemical reactions. How can we automatically design functional enzymes? In this paper, we propose EnzyGen, an approach to learn a unified model to design enzymes across all functional families. Our key idea is to generate an enzyme's amino acid sequence and their three-dimensional (3D) coordinates based on functionally important sites and substrates corresponding to a desired catalytic function. These sites are automatically mined from enzyme databases. EnzyGen consists of a novel interleaving network of attention and neighborhood equivariant layers, which captures both long-range correlation in an entire protein sequence and local influence from nearest amino acids in 3D space. To learn the generative model, we devise a joint training objective, including a sequence generation loss, a position prediction loss and an enzyme-substrate interaction loss. We further construct EnzyBench, a dataset with 3157 enzyme families, covering all available enzymes within the protein data bank (PDB). Experimental results show that our EnzyGen consistently achieves the best performance across all 323 testing families, surpassing the best baseline by 10.79% in terms of substrate binding affinity. These findings demonstrate EnzyGen's superior capability in designing well-folded and effective enzymes binding to specific substrates with high affinities.
Functional Geometry Guided Protein Sequence and Backbone Structure Co-Design
Oct 09, 2023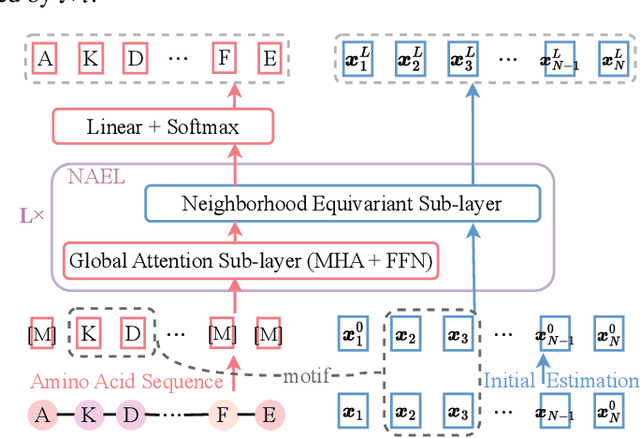
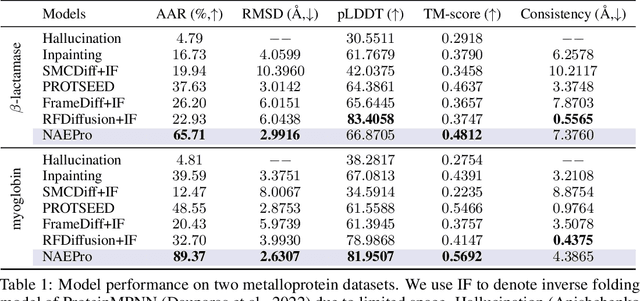
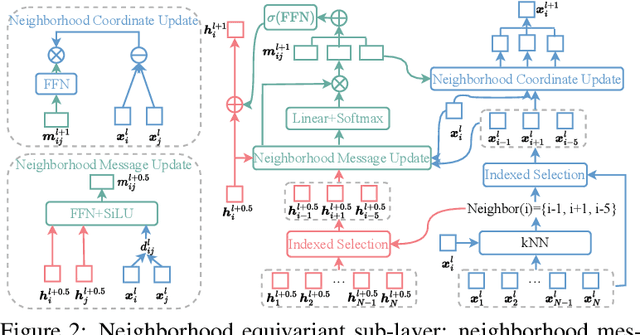
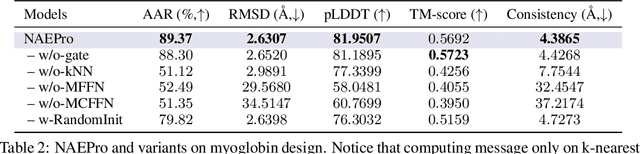
Proteins are macromolecules responsible for essential functions in almost all living organisms. Designing reasonable proteins with desired functions is crucial. A protein's sequence and structure are strongly correlated and they together determine its function. In this paper, we propose NAEPro, a model to jointly design Protein sequence and structure based on automatically detected functional sites. NAEPro is powered by an interleaving network of attention and equivariant layers, which can capture global correlation in a whole sequence and local influence from nearest amino acids in three dimensional (3D) space. Such an architecture facilitates effective yet economic message passing at two levels. We evaluate our model and several strong baselines on two protein datasets, $\beta$-lactamase and myoglobin. Experimental results show that our model consistently achieves the highest amino acid recovery rate, TM-score, and the lowest RMSD among all competitors. These findings prove the capability of our model to design protein sequences and structures that closely resemble their natural counterparts. Furthermore, in-depth analysis further confirms our model's ability to generate highly effective proteins capable of binding to their target metallocofactors. We provide code, data and models in Github.
Joint Design of Protein Sequence and Structure based on Motifs
Oct 04, 2023Designing novel proteins with desired functions is crucial in biology and chemistry. However, most existing work focus on protein sequence design, leaving protein sequence and structure co-design underexplored. In this paper, we propose GeoPro, a method to design protein backbone structure and sequence jointly. Our motivation is that protein sequence and its backbone structure constrain each other, and thus joint design of both can not only avoid nonfolding and misfolding but also produce more diverse candidates with desired functions. To this end, GeoPro is powered by an equivariant encoder for three-dimensional (3D) backbone structure and a protein sequence decoder guided by 3D geometry. Experimental results on two biologically significant metalloprotein datasets, including $\beta$-lactamases and myoglobins, show that our proposed GeoPro outperforms several strong baselines on most metrics. Remarkably, our method discovers novel $\beta$-lactamases and myoglobins which are not present in protein data bank (PDB) and UniProt. These proteins exhibit stable folding and active site environments reminiscent of those of natural proteins, demonstrating their excellent potential to be biologically functional.
Follow Your Path: a Progressive Method for Knowledge Distillation
Jul 20, 2021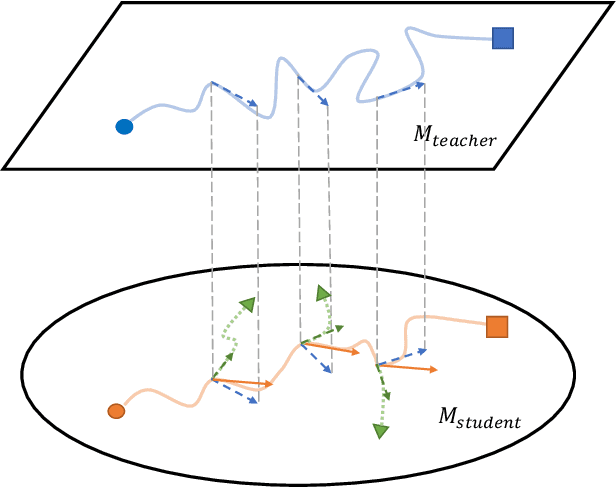


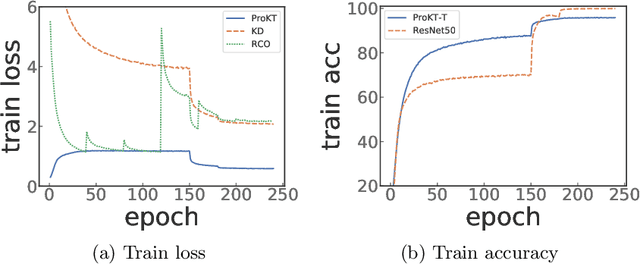
Deep neural networks often have a huge number of parameters, which posts challenges in deployment in application scenarios with limited memory and computation capacity. Knowledge distillation is one approach to derive compact models from bigger ones. However, it has been observed that a converged heavy teacher model is strongly constrained for learning a compact student network and could make the optimization subject to poor local optima. In this paper, we propose ProKT, a new model-agnostic method by projecting the supervision signals of a teacher model into the student's parameter space. Such projection is implemented by decomposing the training objective into local intermediate targets with an approximate mirror descent technique. The proposed method could be less sensitive with the quirks during optimization which could result in a better local optimum. Experiments on both image and text datasets show that our proposed ProKT consistently achieves superior performance compared to other existing knowledge distillation methods.
Variational Template Machine for Data-to-Text Generation
Feb 13, 2020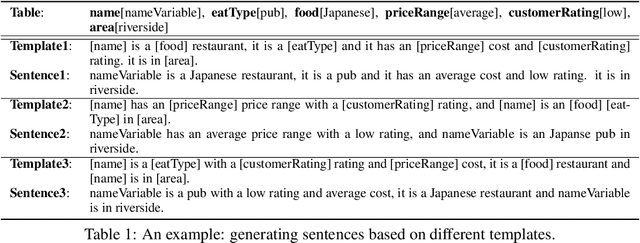
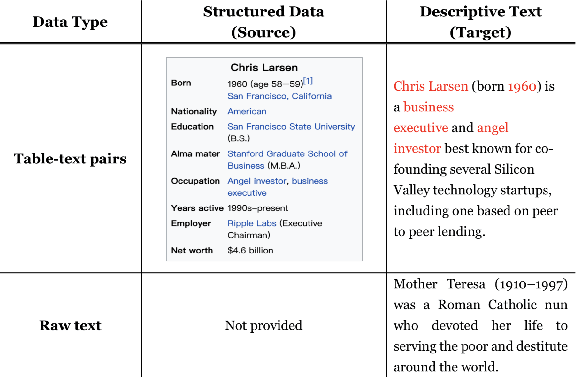


How to generate descriptions from structured data organized in tables? Existing approaches using neural encoder-decoder models often suffer from lacking diversity. We claim that an open set of templates is crucial for enriching the phrase constructions and realizing varied generations. Learning such templates is prohibitive since it often requires a large paired <table, description> corpus, which is seldom available. This paper explores the problem of automatically learning reusable "templates" from paired and non-paired data. We propose the variational template machine (VTM), a novel method to generate text descriptions from data tables. Our contributions include: a) we carefully devise a specific model architecture and losses to explicitly disentangle text template and semantic content information, in the latent spaces, and b)we utilize both small parallel data and large raw text without aligned tables to enrich the template learning. Experiments on datasets from a variety of different domains show that VTM is able to generate more diversely while keeping a good fluency and quality.
Kernelized Bayesian Softmax for Text Generation
Nov 01, 2019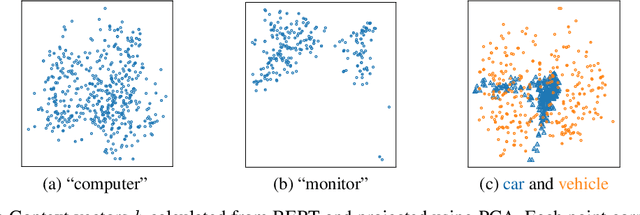
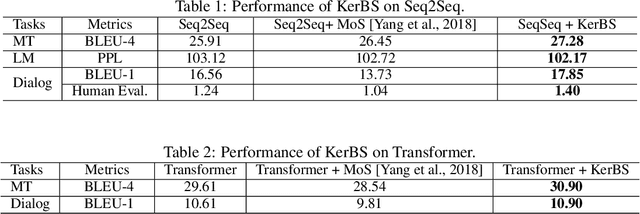
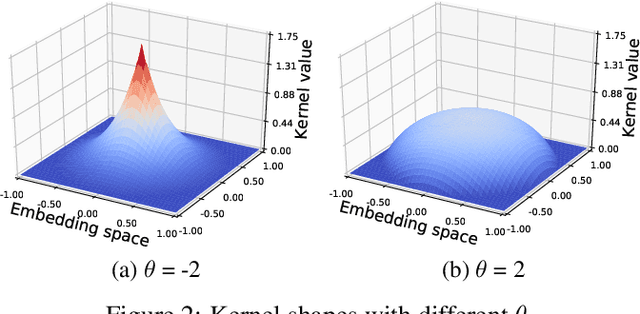

Neural models for text generation require a softmax layer with proper token embeddings during the decoding phase. Most existing approaches adopt single point embedding for each token. However, a word may have multiple senses according to different context, some of which might be distinct. In this paper, we propose KerBS, a novel approach for learning better embeddings for text generation. KerBS embodies two advantages: (a) it employs a Bayesian composition of embeddings for words with multiple senses; (b) it is adaptive to semantic variances of words and robust to rare sentence context by imposing learned kernels to capture the closeness of words (senses) in the embedding space. Empirical studies show that KerBS significantly boosts the performance of several text generation tasks.
Fixing Gaussian Mixture VAEs for Interpretable Text Generation
Jun 16, 2019
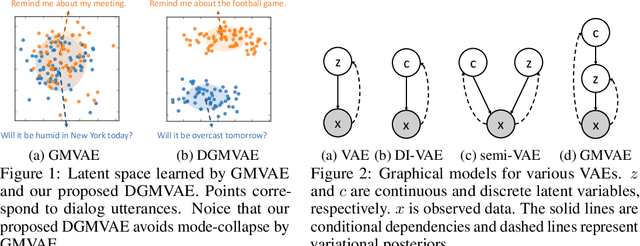


Variational auto-encoder (VAE) with Gaussian priors is effective in text generation. To improve the controllability and interpretability, we propose to use Gaussian mixture distribution as the prior for VAE (GMVAE), since it includes an extra discrete latent variable in addition to the continuous one. Unfortunately, training GMVAE using standard variational approximation often leads to the mode-collapse problem. We theoretically analyze the root cause --- maximizing the evidence lower bound of GMVAE implicitly aggregates the means of multiple Gaussian priors. We propose Dispersed-GMVAE (DGMVAE), an improved model for text generation. It introduces two extra terms to alleviate mode-collapse and to induce a better structured latent space. Experimental results show that DGMVAE outperforms strong baselines in several language modeling and text generation benchmarks.