 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProxelGen: Generating Proteins as 3D Densities
Jun 24, 2025We develop ProxelGen, a protein structure generative model that operates on 3D densities as opposed to the prevailing 3D point cloud representations. Representing proteins as voxelized densities, or proxels, enables new tasks and conditioning capabilities. We generate proteins encoded as proxels via a 3D CNN-based VAE in conjunction with a diffusion model operating on its latent space. Compared to state-of-the-art models, ProxelGen's samples achieve higher novelty, better FID scores, and the same level of designability as the training set. ProxelGen's advantages are demonstrated in a standard motif scaffolding benchmark, and we show how 3D density-based generation allows for more flexible shape conditioning.
Predicting sub-population specific viral evolution
Oct 28, 2024



Forecasting the change in the distribution of viral variants is crucial for therapeutic design and disease surveillance. This task poses significant modeling challenges due to the sharp differences in virus distributions across sub-populations (e.g., countries) and their dynamic interactions. Existing machine learning approaches that model the variant distribution as a whole are incapable of making location-specific predictions and ignore transmissions that shape the viral landscape. In this paper, we propose a sub-population specific protein evolution model, which predicts the time-resolved distributions of viral proteins in different locations. The algorithm explicitly models the transmission rates between sub-populations and learns their interdependence from data. The change in protein distributions across all sub-populations is defined through a linear ordinary differential equation (ODE) parametrized by transmission rates. Solving this ODE yields the likelihood of a given protein occurring in particular sub-populations. Multi-year evaluation on both SARS-CoV-2 and influenza A/H3N2 demonstrates that our model outperforms baselines in accurately predicting distributions of viral proteins across continents and countries. We also find that the transmission rates learned from data are consistent with the transmission pathways discovered by retrospective phylogenetic analysis.
Predicting perturbation targets with causal differential networks
Oct 04, 2024



Rationally identifying variables responsible for changes to a biological system can enable myriad applications in disease understanding and cell engineering. From a causality perspective, we are given two datasets generated by the same causal model, one observational (control) and one interventional (perturbed). The goal is to isolate the subset of measured variables (e.g. genes) that were the targets of the intervention, i.e. those whose conditional independencies have changed. Knowing the causal graph would limit the search space, allowing us to efficiently pinpoint these variables. However, current algorithms that infer causal graphs in the presence of unknown intervention targets scale poorly to the hundreds or thousands of variables in biological data, as they must jointly search the combinatorial spaces of graphs and consistent intervention targets. In this work, we propose a causality-inspired approach for predicting perturbation targets that decouples the two search steps. First, we use an amortized causal discovery model to separately infer causal graphs from the observational and interventional datasets. Then, we learn to map these paired graphs to the sets of variables that were intervened upon, in a supervised learning framework. This approach consistently outperforms baselines for perturbation modeling on seven single-cell transcriptomics datasets, each with thousands of measured variables. We also demonstrate significant improvements over six causal discovery algorithms in predicting intervention targets across a variety of tractable, synthetic datasets.
OpenChemIE: An Information Extraction Toolkit For Chemistry Literature
Apr 01, 2024



Information extraction from chemistry literature is vital for constructing up-to-date reaction databases for data-driven chemistry. Complete extraction requires combining information across text, tables, and figures, whereas prior work has mainly investigated extracting reactions from single modalities. In this paper, we present OpenChemIE to address this complex challenge and enable the extraction of reaction data at the document level. OpenChemIE approaches the problem in two steps: extracting relevant information from individual modalities and then integrating the results to obtain a final list of reactions. For the first step, we employ specialized neural models that each address a specific task for chemistry information extraction, such as parsing molecules or reactions from text or figures. We then integrate the information from these modules using chemistry-informed algorithms, allowing for the extraction of fine-grained reaction data from reaction condition and substrate scope investigations. Our machine learning models attain state-of-the-art performance when evaluated individually, and we meticulously annotate a challenging dataset of reaction schemes with R-groups to evaluate our pipeline as a whole, achieving an F1 score of 69.5%. Additionally, the reaction extraction results of \ours attain an accuracy score of 64.3% when directly compared against the Reaxys chemical database. We provide OpenChemIE freely to the public as an open-source package, as well as through a web interface.
Deep Confident Steps to New Pockets: Strategies for Docking Generalization
Feb 28, 2024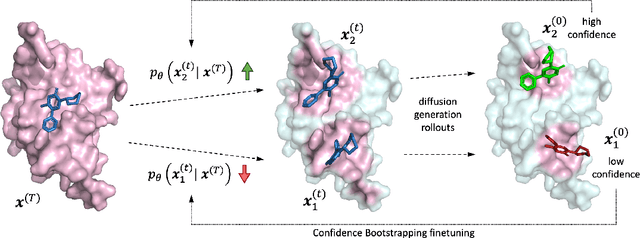
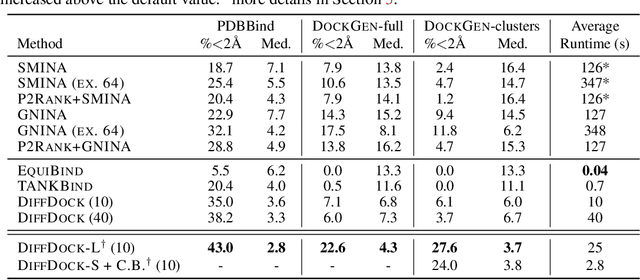
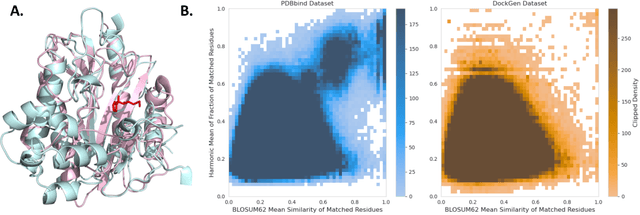
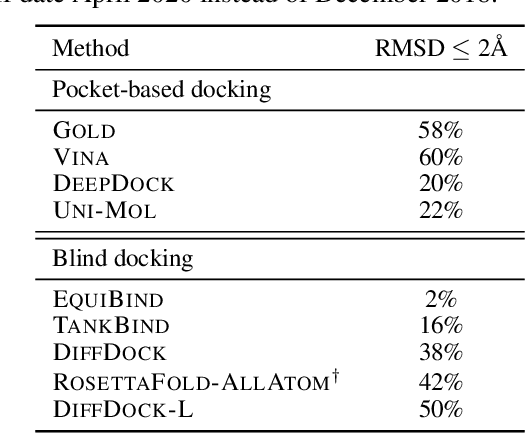
Accurate blind docking has the potential to lead to new biological breakthroughs, but for this promise to be realized, docking methods must generalize well across the proteome. Existing benchmarks, however, fail to rigorously assess generalizability. Therefore, we develop DockGen, a new benchmark based on the ligand-binding domains of proteins, and we show that existing machine learning-based docking models have very weak generalization abilities. We carefully analyze the scaling laws of ML-based docking and show that, by scaling data and model size, as well as integrating synthetic data strategies, we are able to significantly increase the generalization capacity and set new state-of-the-art performance across benchmarks. Further, we propose Confidence Bootstrapping, a new training paradigm that solely relies on the interaction between diffusion and confidence models and exploits the multi-resolution generation process of diffusion models. We demonstrate that Confidence Bootstrapping significantly improves the ability of ML-based docking methods to dock to unseen protein classes, edging closer to accurate and generalizable blind docking methods.
Dirichlet Flow Matching with Applications to DNA Sequence Design
Feb 08, 2024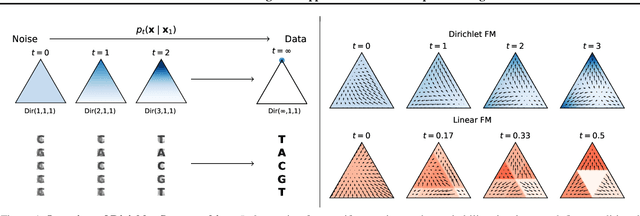

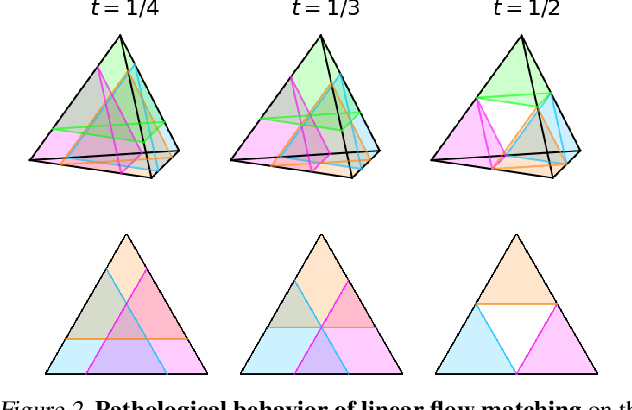
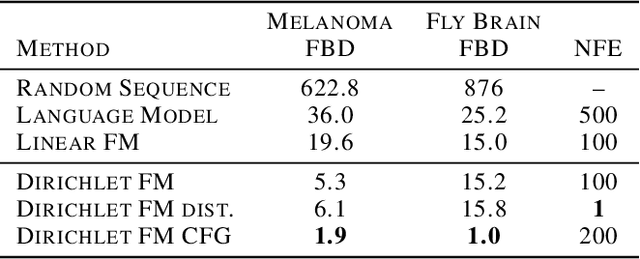
Discrete diffusion or flow models could enable faster and more controllable sequence generation than autoregressive models. We show that na\"ive linear flow matching on the simplex is insufficient toward this goal since it suffers from discontinuities in the training target and further pathologies. To overcome this, we develop Dirichlet flow matching on the simplex based on mixtures of Dirichlet distributions as probability paths. In this framework, we derive a connection between the mixtures' scores and the flow's vector field that allows for classifier and classifier-free guidance. Further, we provide distilled Dirichlet flow matching, which enables one-step sequence generation with minimal performance hits, resulting in $O(L)$ speedups compared to autoregressive models. On complex DNA sequence generation tasks, we demonstrate superior performance compared to all baselines in distributional metrics and in achieving desired design targets for generated sequences. Finally, we show that our classifier-free guidance approach improves unconditional generation and is effective for generating DNA that satisfies design targets. Code is available at https://github.com/HannesStark/dirichlet-flow-matching.
Generative Flows on Discrete State-Spaces: Enabling Multimodal Flows with Applications to Protein Co-Design
Feb 07, 2024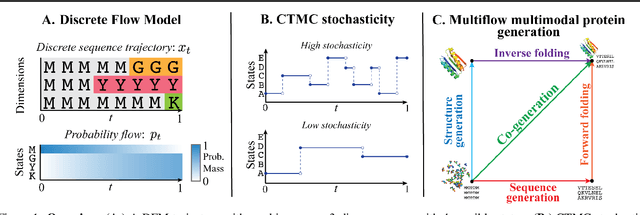



Combining discrete and continuous data is an important capability for generative models. We present Discrete Flow Models (DFMs), a new flow-based model of discrete data that provides the missing link in enabling flow-based generative models to be applied to multimodal continuous and discrete data problems. Our key insight is that the discrete equivalent of continuous space flow matching can be realized using Continuous Time Markov Chains. DFMs benefit from a simple derivation that includes discrete diffusion models as a specific instance while allowing improved performance over existing diffusion-based approaches. We utilize our DFMs method to build a multimodal flow-based modeling framework. We apply this capability to the task of protein co-design, wherein we learn a model for jointly generating protein structure and sequence. Our approach achieves state-of-the-art co-design performance while allowing the same multimodal model to be used for flexible generation of the sequence or structure.
Sample, estimate, aggregate: A recipe for causal discovery foundation models
Feb 02, 2024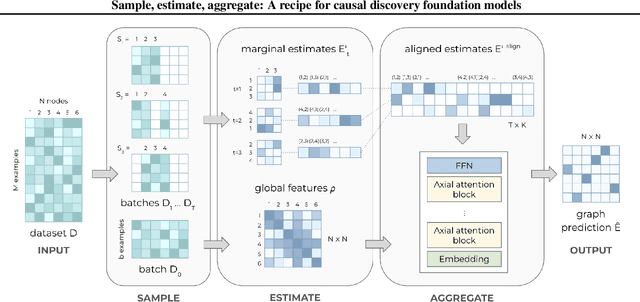

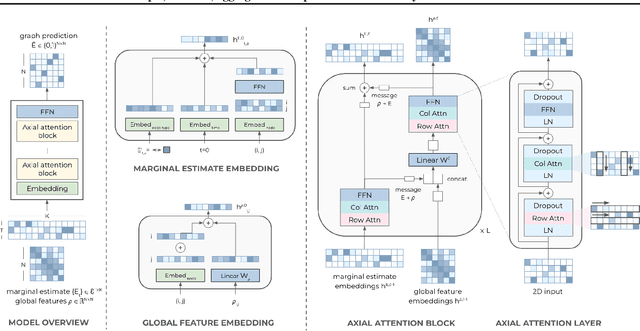
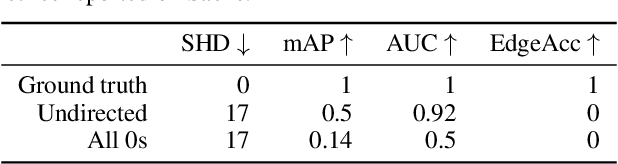
Causal discovery, the task of inferring causal structure from data, promises to accelerate scientific research, inform policy making, and more. However, the per-dataset nature of existing causal discovery algorithms renders them slow, data hungry, and brittle. Inspired by foundation models, we propose a causal discovery framework where a deep learning model is pretrained to resolve predictions from classical discovery algorithms run over smaller subsets of variables. This method is enabled by the observations that the outputs from classical algorithms are fast to compute for small problems, informative of (marginal) data structure, and their structure outputs as objects remain comparable across datasets. Our method achieves state-of-the-art performance on synthetic and realistic datasets, generalizes to data generating mechanisms not seen during training, and offers inference speeds that are orders of magnitude faster than existing models.
Improved motif-scaffolding with SE(3) flow matching
Jan 08, 2024Protein design often begins with knowledge of a desired function from a motif which motif-scaffolding aims to construct a functional protein around. Recently, generative models have achieved breakthrough success in designing scaffolds for a diverse range of motifs. However, the generated scaffolds tend to lack structural diversity, which can hinder success in wet-lab validation. In this work, we extend FrameFlow, an SE(3) flow matching model for protein backbone generation, to perform motif-scaffolding with two complementary approaches. The first is motif amortization, in which FrameFlow is trained with the motif as input using a data augmentation strategy. The second is motif guidance, which performs scaffolding using an estimate of the conditional score from FrameFlow, and requires no additional training. Both approaches achieve an equivalent or higher success rate than previous state-of-the-art methods, with 2.5 times more structurally diverse scaffolds. Code: https://github.com/ microsoft/frame-flow.
Predictive Chemistry Augmented with Text Retrieval
Dec 08, 2023



This paper focuses on using natural language descriptions to enhance predictive models in the chemistry field. Conventionally, chemoinformatics models are trained with extensive structured data manually extracted from the literature. In this paper, we introduce TextReact, a novel method that directly augments predictive chemistry with texts retrieved from the literature. TextReact retrieves text descriptions relevant for a given chemical reaction, and then aligns them with the molecular representation of the reaction. This alignment is enhanced via an auxiliary masked LM objective incorporated in the predictor training. We empirically validate the framework on two chemistry tasks: reaction condition recommendation and one-step retrosynthesis. By leveraging text retrieval, TextReact significantly outperforms state-of-the-art chemoinformatics models trained solely on molecular data.