 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeASKCOS: an open source software suite for synthesis planning
Jan 03, 2025



The advancement of machine learning and the availability of large-scale reaction datasets have accelerated the development of data-driven models for computer-aided synthesis planning (CASP) in the past decade. Here, we detail the newest version of ASKCOS, an open source software suite for synthesis planning that makes available several research advances in a freely available, practical tool. Four one-step retrosynthesis models form the basis of both interactive planning and automatic planning modes. Retrosynthetic planning is complemented by other modules for feasibility assessment and pathway evaluation, including reaction condition recommendation, reaction outcome prediction, and auxiliary capabilities such as solubility prediction and quantum mechanical descriptor prediction. ASKCOS has assisted hundreds of medicinal, synthetic, and process chemists in their day-to-day tasks, complementing expert decision making. It is our belief that CASP tools like ASKCOS are an important part of modern chemistry research, and that they offer ever-increasing utility and accessibility.
SDDBench: A Benchmark for Synthesizable Drug Design
Nov 13, 2024



A significant challenge in wet lab experiments with current drug design generative models is the trade-off between pharmacological properties and synthesizability. Molecules predicted to have highly desirable properties are often difficult to synthesize, while those that are easily synthesizable tend to exhibit less favorable properties. As a result, evaluating the synthesizability of molecules in general drug design scenarios remains a significant challenge in the field of drug discovery. The commonly used synthetic accessibility (SA) score aims to evaluate the ease of synthesizing generated molecules, but it falls short of guaranteeing that synthetic routes can actually be found. Inspired by recent advances in top-down synthetic route generation, we propose a new, data-driven metric to evaluate molecule synthesizability. Our approach directly assesses the feasibility of synthetic routes for a given molecule through our proposed round-trip score. This novel metric leverages the synergistic duality between retrosynthetic planners and reaction predictors, both of which are trained on extensive reaction datasets. To demonstrate the efficacy of our method, we conduct a comprehensive evaluation of round-trip scores alongside search success rate across a range of representative molecule generative models. Code is available at https://github.com/SongtaoLiu0823/SDDBench.
Beyond Major Product Prediction: Reproducing Reaction Mechanisms with Machine Learning Models Trained on a Large-Scale Mechanistic Dataset
Mar 07, 2024
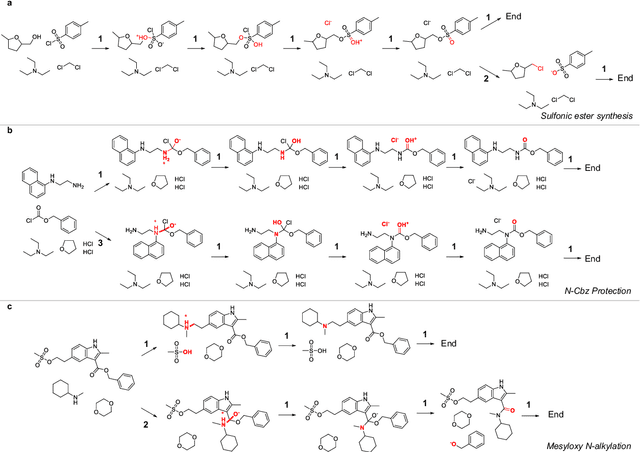
Mechanistic understanding of organic reactions can facilitate reaction development, impurity prediction, and in principle, reaction discovery. While several machine learning models have sought to address the task of predicting reaction products, their extension to predicting reaction mechanisms has been impeded by the lack of a corresponding mechanistic dataset. In this study, we construct such a dataset by imputing intermediates between experimentally reported reactants and products using expert reaction templates and train several machine learning models on the resulting dataset of 5,184,184 elementary steps. We explore the performance and capabilities of these models, focusing on their ability to predict reaction pathways and recapitulate the roles of catalysts and reagents. Additionally, we demonstrate the potential of mechanistic models in predicting impurities, often overlooked by conventional models. We conclude by evaluating the generalizability of mechanistic models to new reaction types, revealing challenges related to dataset diversity, consecutive predictions, and violations of atom conservation.
Predictive Chemistry Augmented with Text Retrieval
Dec 08, 2023



This paper focuses on using natural language descriptions to enhance predictive models in the chemistry field. Conventionally, chemoinformatics models are trained with extensive structured data manually extracted from the literature. In this paper, we introduce TextReact, a novel method that directly augments predictive chemistry with texts retrieved from the literature. TextReact retrieves text descriptions relevant for a given chemical reaction, and then aligns them with the molecular representation of the reaction. This alignment is enhanced via an auxiliary masked LM objective incorporated in the predictor training. We empirically validate the framework on two chemistry tasks: reaction condition recommendation and one-step retrosynthesis. By leveraging text retrieval, TextReact significantly outperforms state-of-the-art chemoinformatics models trained solely on molecular data.
RxnScribe: A Sequence Generation Model for Reaction Diagram Parsing
May 19, 2023Reaction diagram parsing is the task of extracting reaction schemes from a diagram in the chemistry literature. The reaction diagrams can be arbitrarily complex, thus robustly parsing them into structured data is an open challenge. In this paper, we present RxnScribe, a machine learning model for parsing reaction diagrams of varying styles. We formulate this structured prediction task with a sequence generation approach, which condenses the traditional pipeline into an end-to-end model. We train RxnScribe on a dataset of 1,378 diagrams and evaluate it with cross validation, achieving an 80.0% soft match F1 score, with significant improvements over previous models. Our code and data are publicly available at https://github.com/thomas0809/RxnScribe.
Robust Molecular Image Recognition: A Graph Generation Approach
May 28, 2022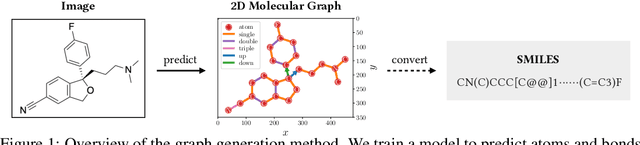
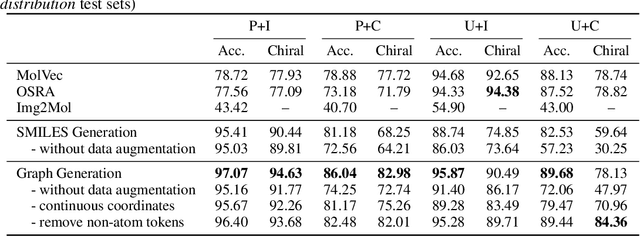
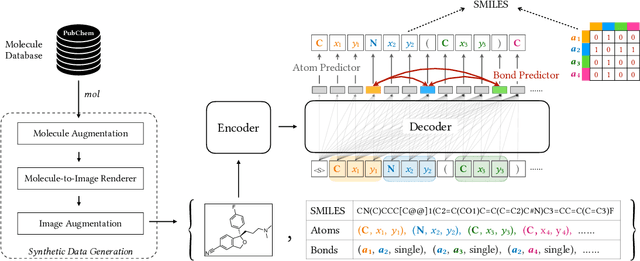
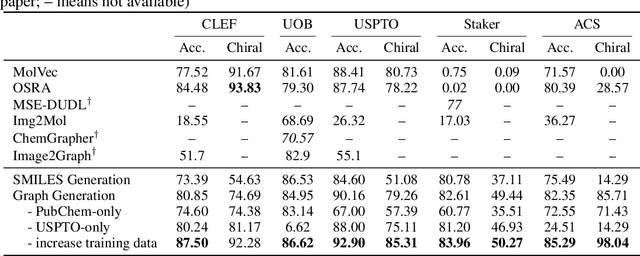
Molecular image recognition is a fundamental task in information extraction from chemistry literature. Previous data-driven models formulate it as an image-to-sequence task, to generate a sequential representation of the molecule (e.g. SMILES string) from its graphical representation. Although they perform adequately on certain benchmarks, these models are not robust in real-world situations, where molecular images differ in style, quality, and chemical patterns. In this paper, we propose a novel graph generation approach that explicitly predicts atoms and bonds, along with their geometric layouts, to construct the molecular graph. We develop data augmentation strategies for molecules and images to increase the robustness of our model against domain shifts. Our model is flexible to incorporate chemistry constraints, and produces more interpretable predictions than SMILES. In experiments on both synthetic and realistic molecular images, our model significantly outperforms previous models, achieving 84-93% accuracy on five benchmarks. We also conduct human evaluation and show that our model reduces the time for a chemist to extract molecular structures from images by roughly 50%.
Permutation invariant graph-to-sequence model for template-free retrosynthesis and reaction prediction
Oct 19, 2021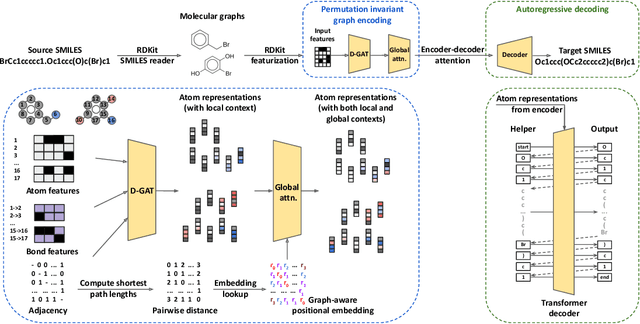
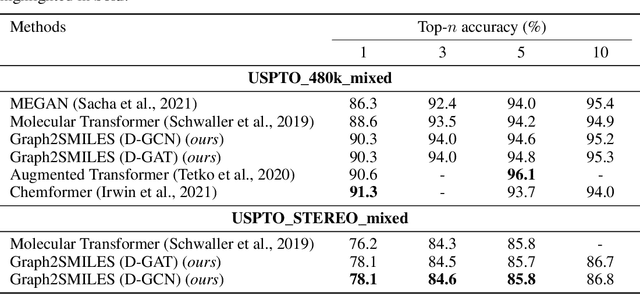
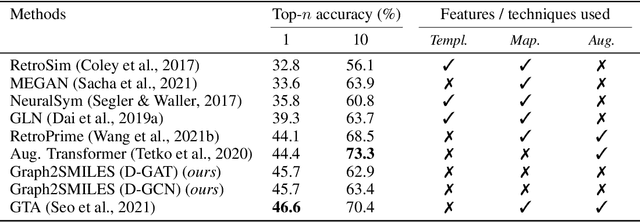
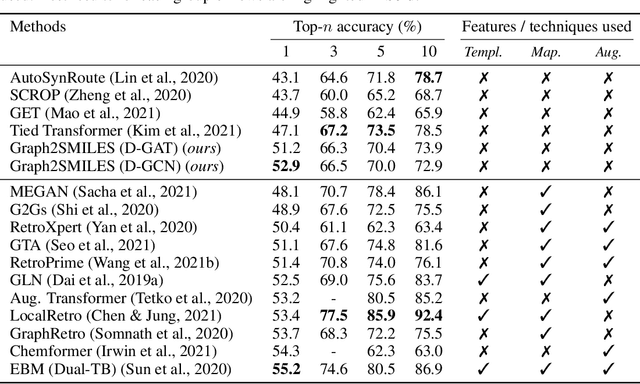
Synthesis planning and reaction outcome prediction are two fundamental problems in computer-aided organic chemistry for which a variety of data-driven approaches have emerged. Natural language approaches that model each problem as a SMILES-to-SMILES translation lead to a simple end-to-end formulation, reduce the need for data preprocessing, and enable the use of well-optimized machine translation model architectures. However, SMILES representations are not an efficient representation for capturing information about molecular structures, as evidenced by the success of SMILES augmentation to boost empirical performance. Here, we describe a novel Graph2SMILES model that combines the power of Transformer models for text generation with the permutation invariance of molecular graph encoders that mitigates the need for input data augmentation. As an end-to-end architecture, Graph2SMILES can be used as a drop-in replacement for the Transformer in any task involving molecule(s)-to-molecule(s) transformations. In our encoder, an attention-augmented directed message passing neural network (D-MPNN) captures local chemical environments, and the global attention encoder allows for long-range and intermolecular interactions, enhanced by graph-aware positional embedding. Graph2SMILES improves the top-1 accuracy of the Transformer baselines by $1.7\%$ and $1.9\%$ for reaction outcome prediction on USPTO_480k and USPTO_STEREO datasets respectively, and by $9.8\%$ for one-step retrosynthesis on the USPTO_50k dataset.
To Paraphrase or Not To Paraphrase: User-Controllable Selective Paraphrase Generation
Aug 21, 2020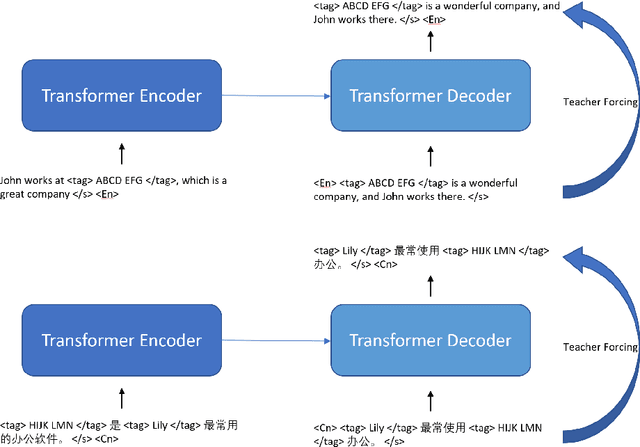
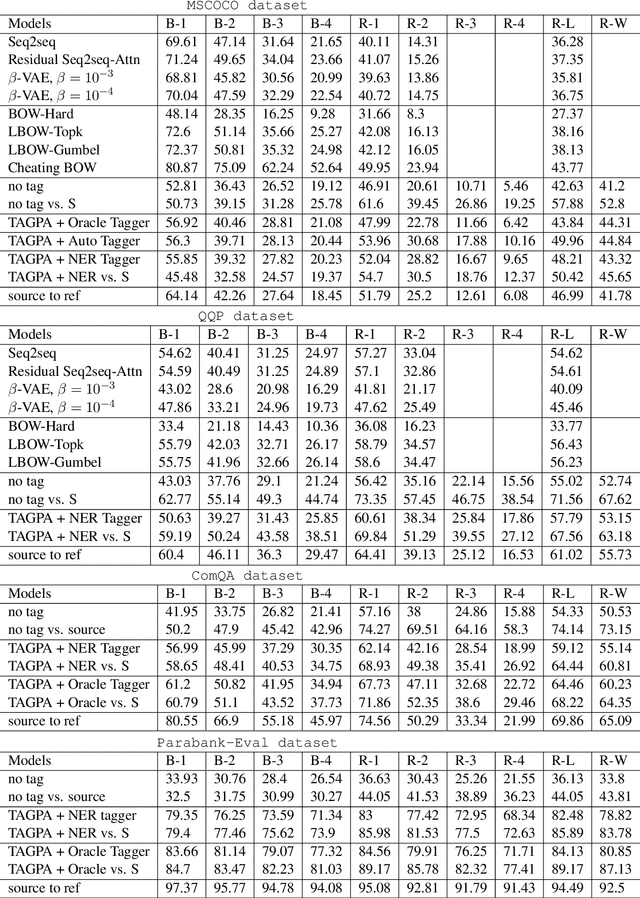
In this article, we propose a paraphrase generation technique to keep the key phrases in source sentences during paraphrasing. We also develop a model called TAGPA with such technique, which has multiple pre-configured or trainable key phrase detector and a paraphrase generator. The paraphrase generator aims to keep the key phrases and increase the diversity of the paraphrased sentences. The key phrases can be entities provided by our user, like company names, people's names, domain-specific terminologies, etc., or can be learned from a given dataset.
Rapid Adaptation of BERT for Information Extraction on Domain-Specific Business Documents
Feb 05, 2020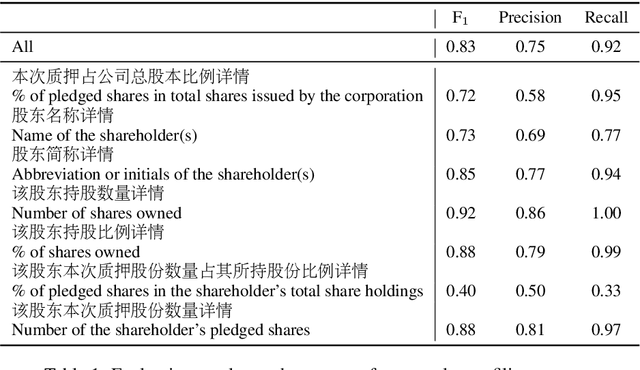
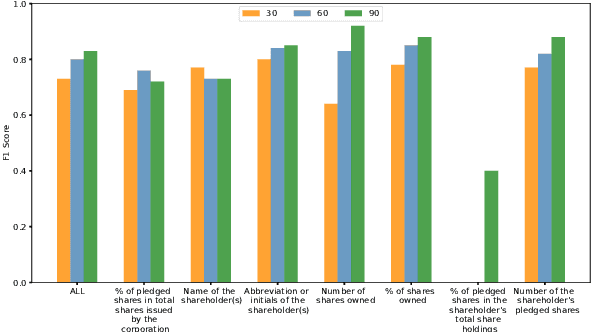
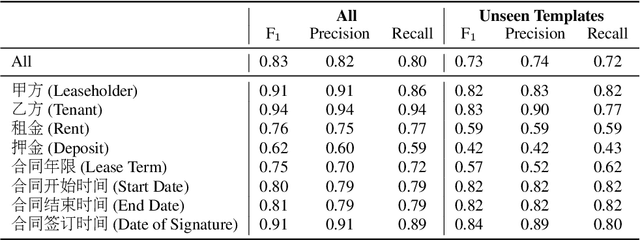
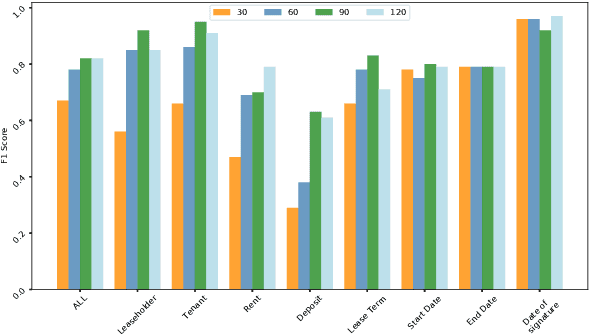
Techniques for automatically extracting important content elements from business documents such as contracts, statements, and filings have the potential to make business operations more efficient. This problem can be formulated as a sequence labeling task, and we demonstrate the adaption of BERT to two types of business documents: regulatory filings and property lease agreements. There are aspects of this problem that make it easier than "standard" information extraction tasks and other aspects that make it more difficult, but on balance we find that modest amounts of annotated data (less than 100 documents) are sufficient to achieve reasonable accuracy. We integrate our models into an end-to-end cloud platform that provides both an easy-to-use annotation interface as well as an inference interface that allows users to upload documents and inspect model outputs.