 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeElectron flow matching for generative reaction mechanism prediction obeying conservation laws
Feb 18, 2025Central to our understanding of chemical reactivity is the principle of mass conservation, which is fundamental for ensuring physical consistency, balancing equations, and guiding reaction design. However, data-driven computational models for tasks such as reaction product prediction rarely abide by this most basic constraint. In this work, we recast the problem of reaction prediction as a problem of electron redistribution using the modern deep generative framework of flow matching. Our model, FlowER, overcomes limitations inherent in previous approaches by enforcing exact mass conservation, thereby resolving hallucinatory failure modes, recovering mechanistic reaction sequences for unseen substrate scaffolds, and generalizing effectively to out-of-domain reaction classes with extremely data-efficient fine-tuning. FlowER additionally enables estimation of thermodynamic or kinetic feasibility and manifests a degree of chemical intuition in reaction prediction tasks. This inherently interpretable framework represents a significant step in bridging the gap between predictive accuracy and mechanistic understanding in data-driven reaction outcome prediction.
ASKCOS: an open source software suite for synthesis planning
Jan 03, 2025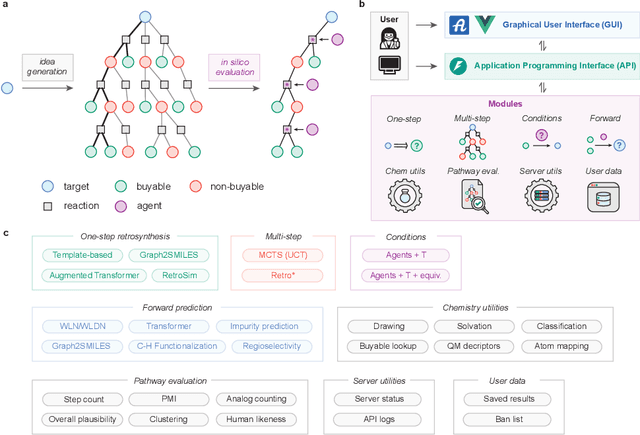



The advancement of machine learning and the availability of large-scale reaction datasets have accelerated the development of data-driven models for computer-aided synthesis planning (CASP) in the past decade. Here, we detail the newest version of ASKCOS, an open source software suite for synthesis planning that makes available several research advances in a freely available, practical tool. Four one-step retrosynthesis models form the basis of both interactive planning and automatic planning modes. Retrosynthetic planning is complemented by other modules for feasibility assessment and pathway evaluation, including reaction condition recommendation, reaction outcome prediction, and auxiliary capabilities such as solubility prediction and quantum mechanical descriptor prediction. ASKCOS has assisted hundreds of medicinal, synthetic, and process chemists in their day-to-day tasks, complementing expert decision making. It is our belief that CASP tools like ASKCOS are an important part of modern chemistry research, and that they offer ever-increasing utility and accessibility.
Beyond Major Product Prediction: Reproducing Reaction Mechanisms with Machine Learning Models Trained on a Large-Scale Mechanistic Dataset
Mar 07, 2024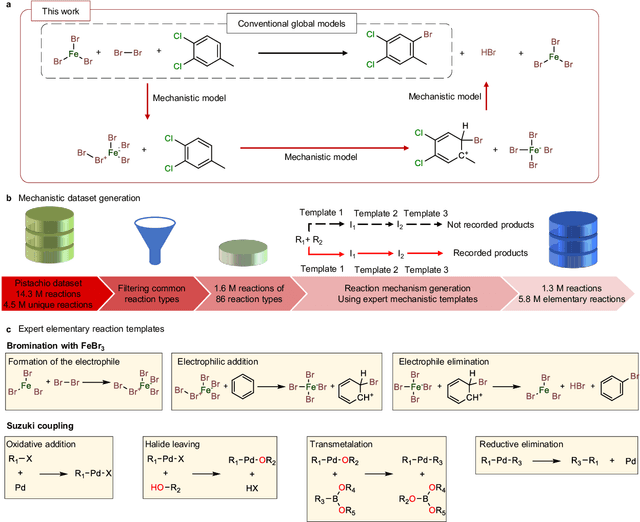
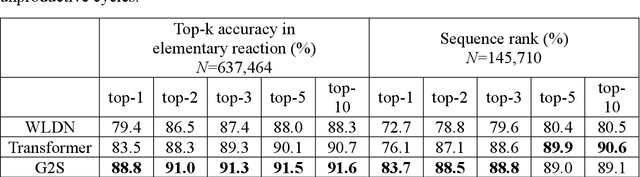
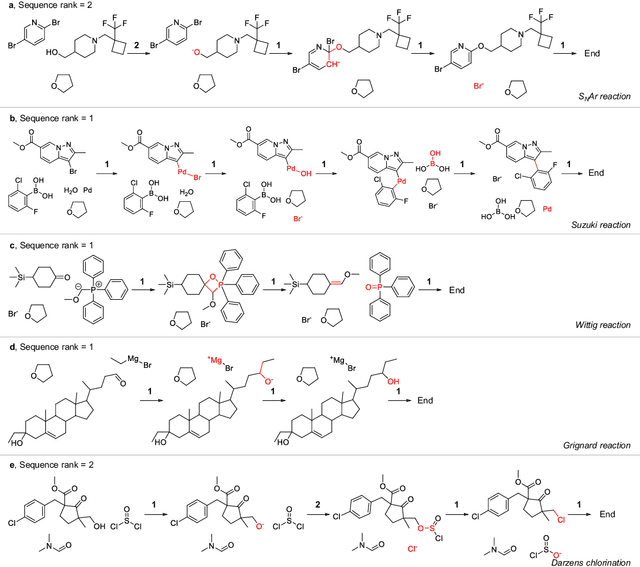
Mechanistic understanding of organic reactions can facilitate reaction development, impurity prediction, and in principle, reaction discovery. While several machine learning models have sought to address the task of predicting reaction products, their extension to predicting reaction mechanisms has been impeded by the lack of a corresponding mechanistic dataset. In this study, we construct such a dataset by imputing intermediates between experimentally reported reactants and products using expert reaction templates and train several machine learning models on the resulting dataset of 5,184,184 elementary steps. We explore the performance and capabilities of these models, focusing on their ability to predict reaction pathways and recapitulate the roles of catalysts and reagents. Additionally, we demonstrate the potential of mechanistic models in predicting impurities, often overlooked by conventional models. We conclude by evaluating the generalizability of mechanistic models to new reaction types, revealing challenges related to dataset diversity, consecutive predictions, and violations of atom conservation.