 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeASKCOS: an open source software suite for synthesis planning
Jan 03, 2025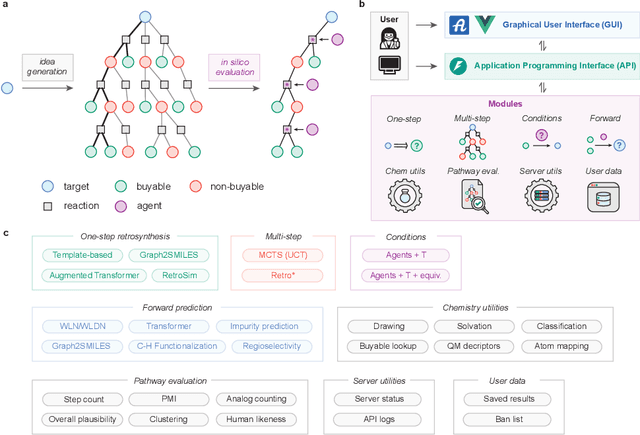



The advancement of machine learning and the availability of large-scale reaction datasets have accelerated the development of data-driven models for computer-aided synthesis planning (CASP) in the past decade. Here, we detail the newest version of ASKCOS, an open source software suite for synthesis planning that makes available several research advances in a freely available, practical tool. Four one-step retrosynthesis models form the basis of both interactive planning and automatic planning modes. Retrosynthetic planning is complemented by other modules for feasibility assessment and pathway evaluation, including reaction condition recommendation, reaction outcome prediction, and auxiliary capabilities such as solubility prediction and quantum mechanical descriptor prediction. ASKCOS has assisted hundreds of medicinal, synthetic, and process chemists in their day-to-day tasks, complementing expert decision making. It is our belief that CASP tools like ASKCOS are an important part of modern chemistry research, and that they offer ever-increasing utility and accessibility.
Double-Ended Synthesis Planning with Goal-Constrained Bidirectional Search
Jul 08, 2024



Computer-aided synthesis planning (CASP) algorithms have demonstrated expert-level abilities in planning retrosynthetic routes to molecules of low to moderate complexity. However, current search methods assume the sufficiency of reaching arbitrary building blocks, failing to address the common real-world constraint where using specific molecules is desired. To this end, we present a formulation of synthesis planning with starting material constraints. Under this formulation, we propose Double-Ended Synthesis Planning (DESP), a novel CASP algorithm under a bidirectional graph search scheme that interleaves expansions from the target and from the goal starting materials to ensure constraint satisfiability. The search algorithm is guided by a goal-conditioned cost network learned offline from a partially observed hypergraph of valid chemical reactions. We demonstrate the utility of DESP in improving solve rates and reducing the number of search expansions by biasing synthesis planning towards expert goals on multiple new benchmarks. DESP can make use of existing one-step retrosynthesis models, and we anticipate its performance to scale as these one-step model capabilities improve.
Beyond Major Product Prediction: Reproducing Reaction Mechanisms with Machine Learning Models Trained on a Large-Scale Mechanistic Dataset
Mar 07, 2024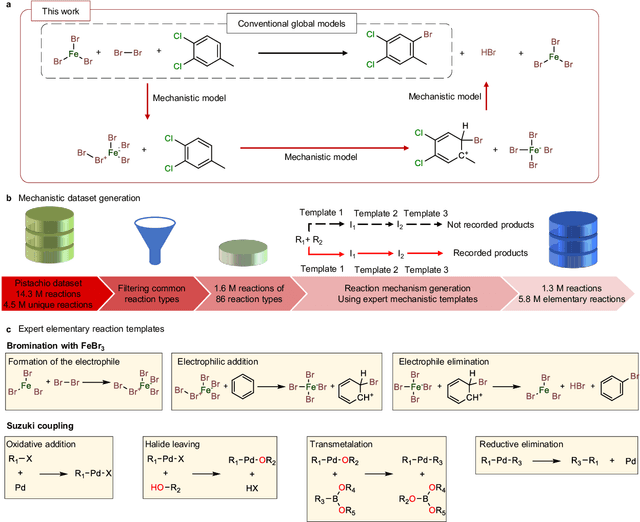
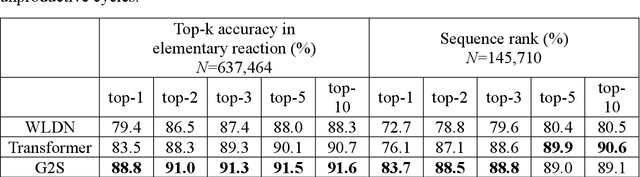
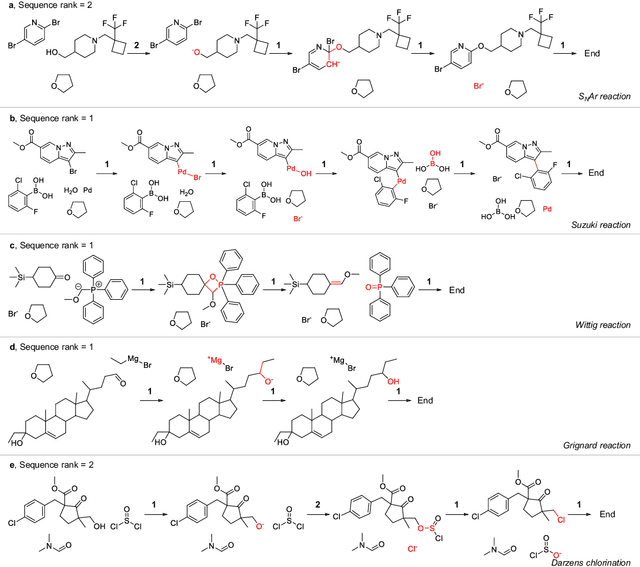
Mechanistic understanding of organic reactions can facilitate reaction development, impurity prediction, and in principle, reaction discovery. While several machine learning models have sought to address the task of predicting reaction products, their extension to predicting reaction mechanisms has been impeded by the lack of a corresponding mechanistic dataset. In this study, we construct such a dataset by imputing intermediates between experimentally reported reactants and products using expert reaction templates and train several machine learning models on the resulting dataset of 5,184,184 elementary steps. We explore the performance and capabilities of these models, focusing on their ability to predict reaction pathways and recapitulate the roles of catalysts and reagents. Additionally, we demonstrate the potential of mechanistic models in predicting impurities, often overlooked by conventional models. We conclude by evaluating the generalizability of mechanistic models to new reaction types, revealing challenges related to dataset diversity, consecutive predictions, and violations of atom conservation.