 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeChallenging reaction prediction models to generalize to novel chemistry
Jan 11, 2025Deep learning models for anticipating the products of organic reactions have found many use cases, including validating retrosynthetic pathways and constraining synthesis-based molecular design tools. Despite compelling performance on popular benchmark tasks, strange and erroneous predictions sometimes ensue when using these models in practice. The core issue is that common benchmarks test models in an in-distribution setting, whereas many real-world uses for these models are in out-of-distribution settings and require a greater degree of extrapolation. To better understand how current reaction predictors work in out-of-distribution domains, we report a series of more challenging evaluations of a prototypical SMILES-based deep learning model. First, we illustrate how performance on randomly sampled datasets is overly optimistic compared to performance when generalizing to new patents or new authors. Second, we conduct time splits that evaluate how models perform when tested on reactions published in years after those in their training set, mimicking real-world deployment. Finally, we consider extrapolation across reaction classes to reflect what would be required for the discovery of novel reaction types. This panel of tasks can reveal the capabilities and limitations of today's reaction predictors, acting as a crucial first step in the development of tomorrow's next-generation models capable of reaction discovery.
Beyond Major Product Prediction: Reproducing Reaction Mechanisms with Machine Learning Models Trained on a Large-Scale Mechanistic Dataset
Mar 07, 2024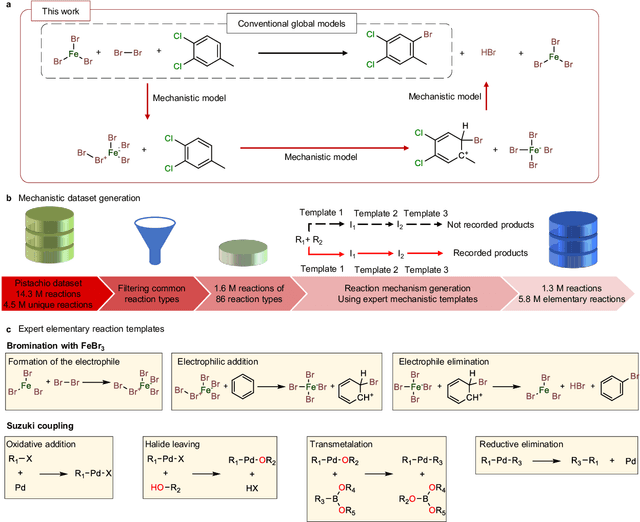
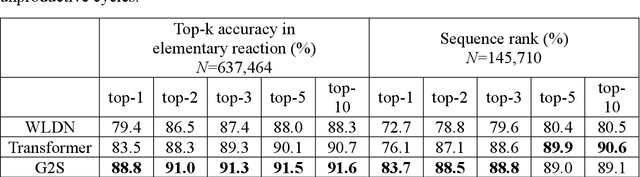
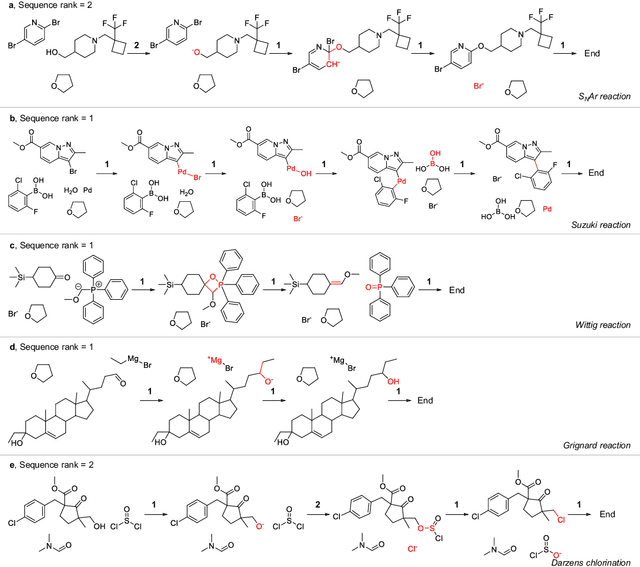
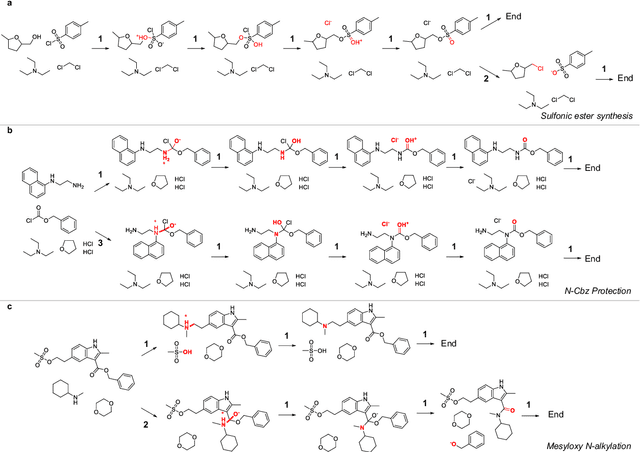
Mechanistic understanding of organic reactions can facilitate reaction development, impurity prediction, and in principle, reaction discovery. While several machine learning models have sought to address the task of predicting reaction products, their extension to predicting reaction mechanisms has been impeded by the lack of a corresponding mechanistic dataset. In this study, we construct such a dataset by imputing intermediates between experimentally reported reactants and products using expert reaction templates and train several machine learning models on the resulting dataset of 5,184,184 elementary steps. We explore the performance and capabilities of these models, focusing on their ability to predict reaction pathways and recapitulate the roles of catalysts and reagents. Additionally, we demonstrate the potential of mechanistic models in predicting impurities, often overlooked by conventional models. We conclude by evaluating the generalizability of mechanistic models to new reaction types, revealing challenges related to dataset diversity, consecutive predictions, and violations of atom conservation.
Prefix-tree Decoding for Predicting Mass Spectra from Molecules
Mar 11, 2023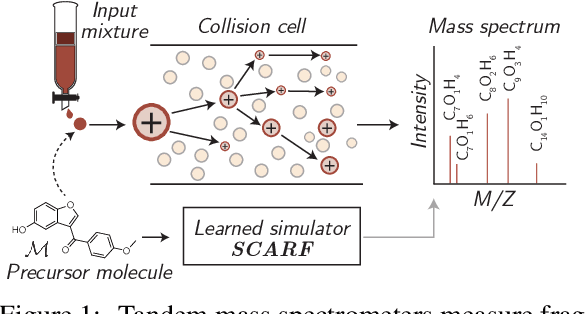
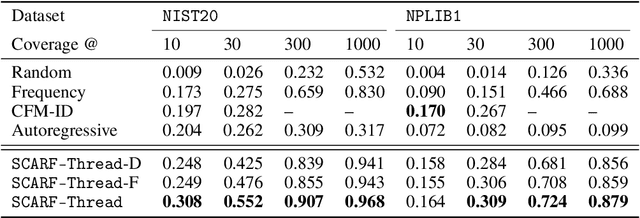
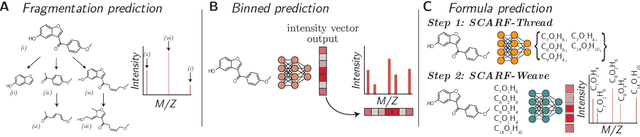
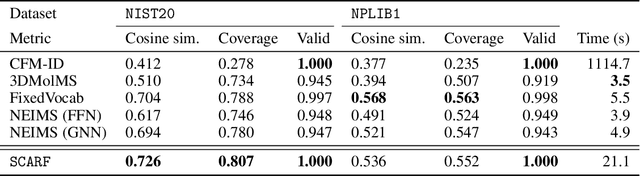
Computational predictions of mass spectra from molecules have enabled the discovery of clinically relevant metabolites. However, such predictive tools are still limited as they occupy one of two extremes, either operating (a) by fragmenting molecules combinatorially with overly rigid constraints on potential rearrangements and poor time complexity or (b) by decoding lossy and nonphysical discretized spectra vectors. In this work, we introduce a new intermediate strategy for predicting mass spectra from molecules by treating mass spectra as sets of chemical formulae, which are themselves multisets of atoms. After first encoding an input molecular graph, we decode a set of chemical subformulae, each of which specify a predicted peak in the mass spectra, the intensities of which are predicted by a second model. Our key insight is to overcome the combinatorial possibilities for chemical subformulae by decoding the formula set using a prefix tree structure, atom-type by atom-type, representing a general method for ordered multiset decoding. We show promising empirical results on mass spectra prediction tasks.
Local Latent Space Bayesian Optimization over Structured Inputs
Jan 28, 2022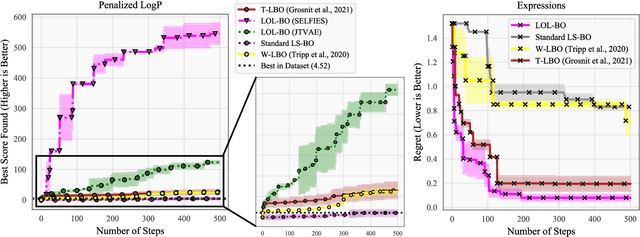
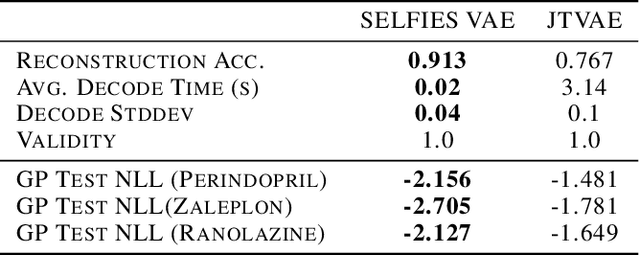
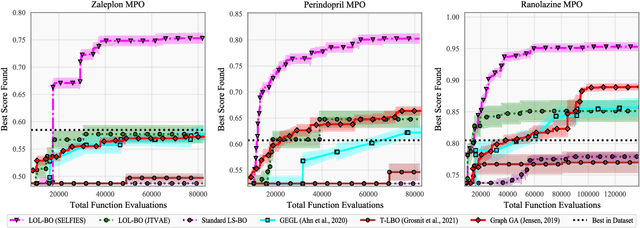
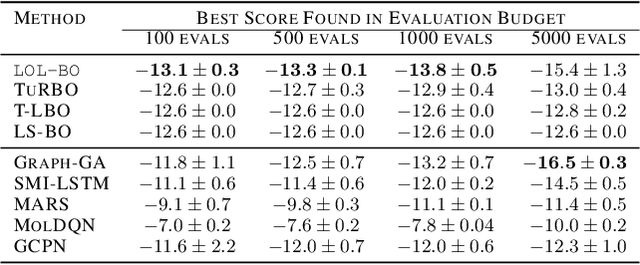
Bayesian optimization over the latent spaces of deep autoencoder models (DAEs) has recently emerged as a promising new approach for optimizing challenging black-box functions over structured, discrete, hard-to-enumerate search spaces (e.g., molecules). Here the DAE dramatically simplifies the search space by mapping inputs into a continuous latent space where familiar Bayesian optimization tools can be more readily applied. Despite this simplification, the latent space typically remains high-dimensional. Thus, even with a well-suited latent space, these approaches do not necessarily provide a complete solution, but may rather shift the structured optimization problem to a high-dimensional one. In this paper, we propose LOL-BO, which adapts the notion of trust regions explored in recent work on high-dimensional Bayesian optimization to the structured setting. By reformulating the encoder to function as both an encoder for the DAE globally and as a deep kernel for the surrogate model within a trust region, we better align the notion of local optimization in the latent space with local optimization in the input space. LOL-BO achieves as much as 20 times improvement over state-of-the-art latent space Bayesian optimization methods across six real-world benchmarks, demonstrating that improvement in optimization strategies is as important as developing better DAE models.
Barking up the right tree: an approach to search over molecule synthesis DAGs
Dec 21, 2020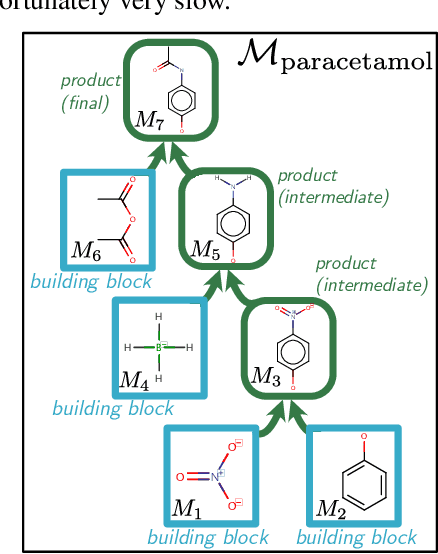
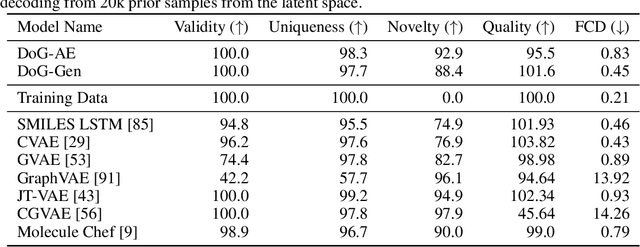
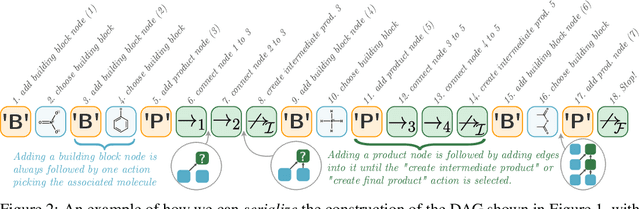
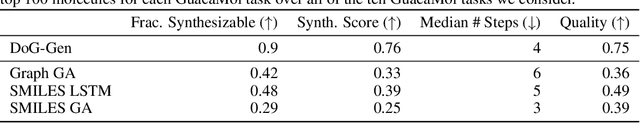
When designing new molecules with particular properties, it is not only important what to make but crucially how to make it. These instructions form a synthesis directed acyclic graph (DAG), describing how a large vocabulary of simple building blocks can be recursively combined through chemical reactions to create more complicated molecules of interest. In contrast, many current deep generative models for molecules ignore synthesizability. We therefore propose a deep generative model that better represents the real world process, by directly outputting molecule synthesis DAGs. We argue that this provides sensible inductive biases, ensuring that our model searches over the same chemical space that chemists would also have access to, as well as interpretability. We show that our approach is able to model chemical space well, producing a wide range of diverse molecules, and allows for unconstrained optimization of an inherently constrained problem: maximize certain chemical properties such that discovered molecules are synthesizable.
A Model to Search for Synthesizable Molecules
Jun 12, 2019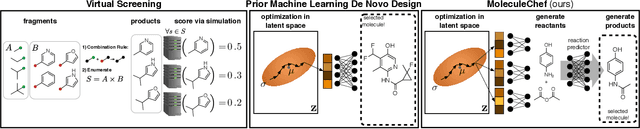
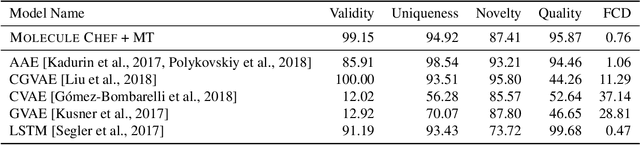
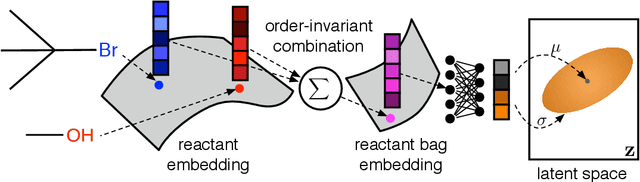
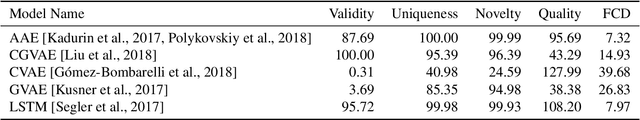
Deep generative models are able to suggest new organic molecules by generating strings, trees, and graphs representing their structure. While such models allow one to generate molecules with desirable properties, they give no guarantees that the molecules can actually be synthesized in practice. We propose a new molecule generation model, mirroring a more realistic real-world process, where (a) reactants are selected, and (b) combined to form more complex molecules. More specifically, our generative model proposes a bag of initial reactants (selected from a pool of commercially-available molecules) and uses a reaction model to predict how they react together to generate new molecules. We first show that the model can generate diverse, valid and unique molecules due to the useful inductive biases of modeling reactions. Furthermore, our model allows chemists to interrogate not only the properties of the generated molecules but also the feasibility of the synthesis routes. We conclude by using our model to solve retrosynthesis problems, predicting a set of reactants that can produce a target product.
Are Generative Classifiers More Robust to Adversarial Attacks?
Jul 09, 2018
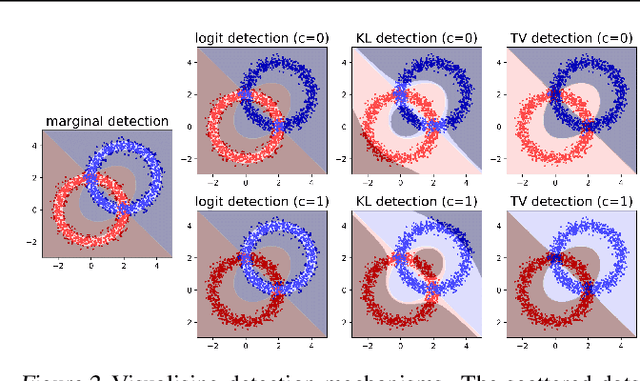
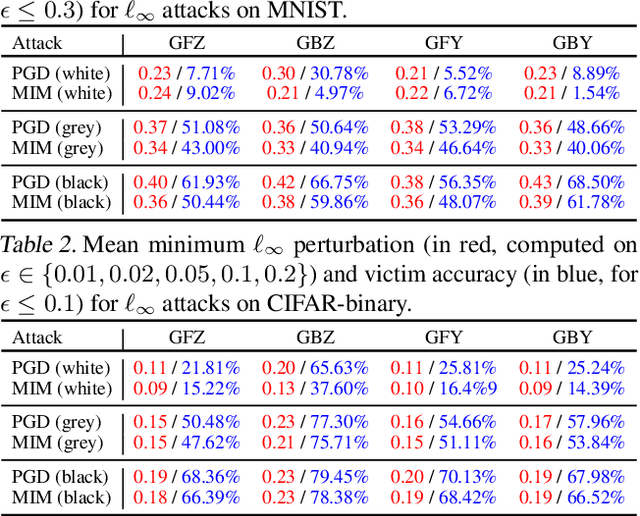
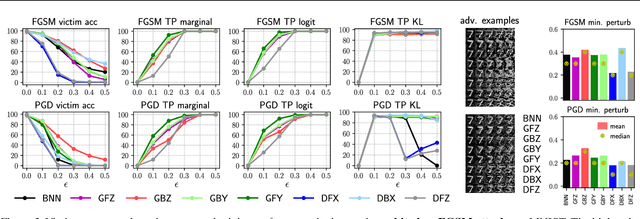
There is a rising interest in studying the robustness of deep neural network classifiers against adversaries, with both advanced attack and defence techniques being actively developed. However, most recent work focuses on discriminative classifiers, which only model the conditional distribution of the labels given the inputs. In this paper we propose the deep Bayes classifier, which improves classical naive Bayes with conditional deep generative models. We further develop detection methods for adversarial examples, which reject inputs that have negative log-likelihood under the generative model exceeding a threshold pre-specified using training data. Experimental results suggest that deep Bayes classifiers are more robust than deep discriminative classifiers, and the proposed detection methods achieve high detection rates against many recently proposed attacks.
Predicting Electron Paths
May 23, 2018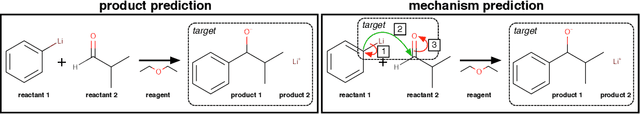
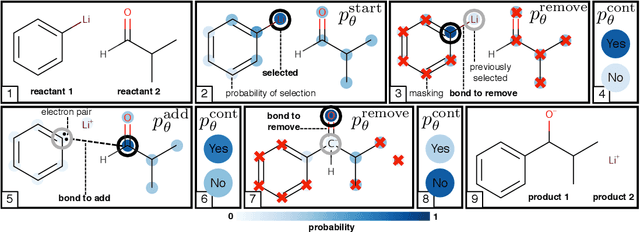
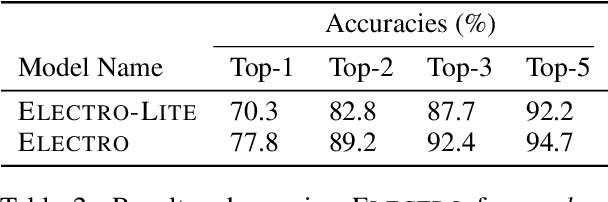
Chemical reactions can be described as the stepwise redistribution of electrons in molecules. As such, reactions are often depicted using "arrow-pushing" diagrams which show this movement as a sequence of arrows. We propose an electron path prediction model (ELECTRO) to learn these sequences directly from raw reaction data. Instead of predicting product molecules directly from reactant molecules in one shot, learning a model of electron movement has the benefits of (a) being easy for chemists to interpret, (b) incorporating constraints of chemistry, such as balanced atom counts before and after the reaction, and (c) naturally encoding the sparsity of chemical reactions, which usually involve changes in only a small number of atoms in the reactants. We design a method to extract approximate reaction paths from any dataset of atom-mapped reaction SMILES strings. Our model achieves state-of-the-art results on a subset of the UPSTO reaction dataset. Furthermore, we show that our model recovers a basic knowledge of chemistry without being explicitly trained to do so.
Adversarial Examples, Uncertainty, and Transfer Testing Robustness in Gaussian Process Hybrid Deep Networks
Jul 08, 2017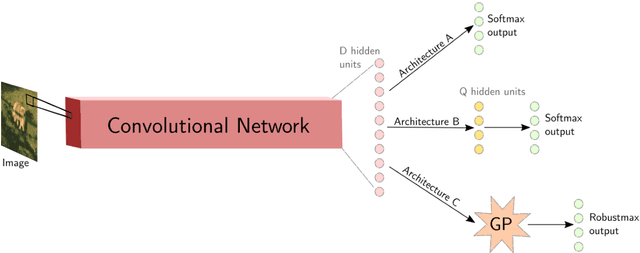
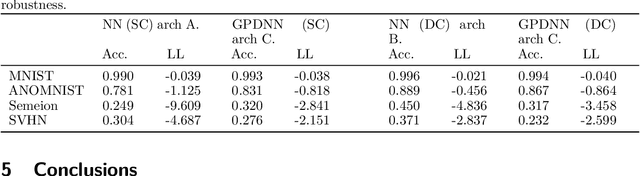
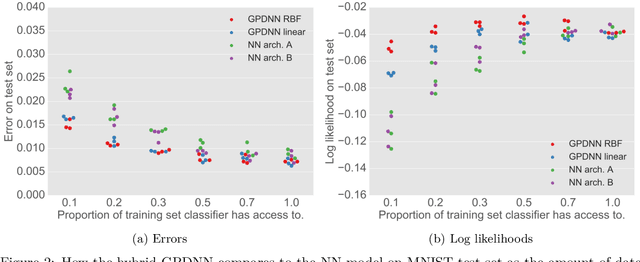
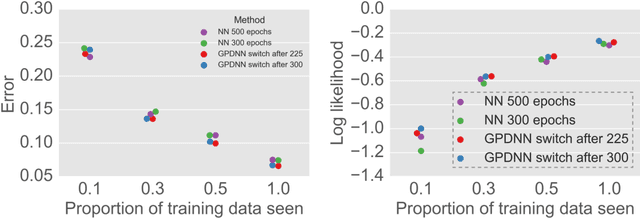
Deep neural networks (DNNs) have excellent representative power and are state of the art classifiers on many tasks. However, they often do not capture their own uncertainties well making them less robust in the real world as they overconfidently extrapolate and do not notice domain shift. Gaussian processes (GPs) with RBF kernels on the other hand have better calibrated uncertainties and do not overconfidently extrapolate far from data in their training set. However, GPs have poor representational power and do not perform as well as DNNs on complex domains. In this paper we show that GP hybrid deep networks, GPDNNs, (GPs on top of DNNs and trained end-to-end) inherit the nice properties of both GPs and DNNs and are much more robust to adversarial examples. When extrapolating to adversarial examples and testing in domain shift settings, GPDNNs frequently output high entropy class probabilities corresponding to essentially "don't know". GPDNNs are therefore promising as deep architectures that know when they don't know.