 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUCS: Estimating Unseen Coverage for Improved In-Context Learning
Apr 13, 2026In-context learning (ICL) performance depends critically on which demonstrations are placed in the prompt, yet most existing selectors prioritize heuristic notions of relevance or diversity and provide limited insight into the coverage of a demonstration set. We propose Unseen Coverage Selection (UKS), a training-free, subset-level coverage prior motivated by the principle that a good demonstration set should expose the model to latent cluster unrevealed by the currently selected subset. UCS operationalizes this idea by (1) inducing discrete latent clusters from model-consistent embeddings and (2) estimating the number of unrevealed clusters within a candidate subset via a Smoothed Good--Turing estimator from its empirical frequency spectrum. Unlike previous selection methods, UCS is coverage-based and training-free, and can be seamlessly combined with both query-dependent and query-independent selection baselines via a simple regularized objective. Experiments on multiple intent-classification and reasoning benchmarks with frontier Large Language Models show that augmenting strong baselines with UCS consistently improves ICL accuracy by up to 2-6% under the same selection budget, while also yielding insights into task- and model-level latent cluster distributions. Code is available at https://github.com/Raina-Xin/UCS.
CAMEL: An ECG Language Model for Forecasting Cardiac Events
Feb 17, 2026Electrocardiograms (ECG) are electrical recordings of the heart that are critical for diagnosing cardiovascular conditions. ECG language models (ELMs) have recently emerged as a promising framework for ECG classification accompanied by report generation. However, current models cannot forecast future cardiac events despite the immense clinical value for planning earlier intervention. To address this gap, we propose CAMEL, the first ELM that is capable of inference over longer signal durations which enables its forecasting capability. Our key insight is a specialized ECG encoder which enables cross-understanding of ECG signals with text. We train CAMEL using established LLM training procedures, combining LoRA adaptation with a curriculum learning pipeline. Our curriculum includes ECG classification, metrics calculations, and multi-turn conversations to elicit reasoning. CAMEL demonstrates strong zero-shot performance across 6 tasks and 9 datasets, including ECGForecastBench, a new benchmark that we introduce for forecasting arrhythmias. CAMEL is on par with or surpasses ELMs and fully supervised baselines both in- and out-of-distribution, achieving SOTA results on ECGBench (+7.0% absolute average gain) as well as ECGForecastBench (+12.4% over fully supervised models and +21.1% over zero-shot ELMs).
I2MoE: Interpretable Multimodal Interaction-aware Mixture-of-Experts
May 25, 2025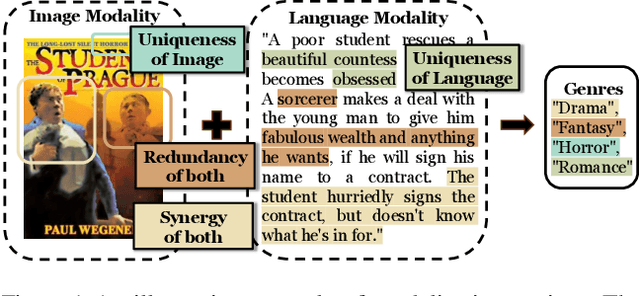
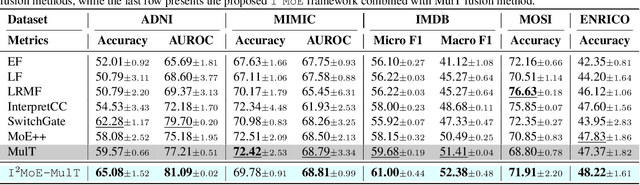

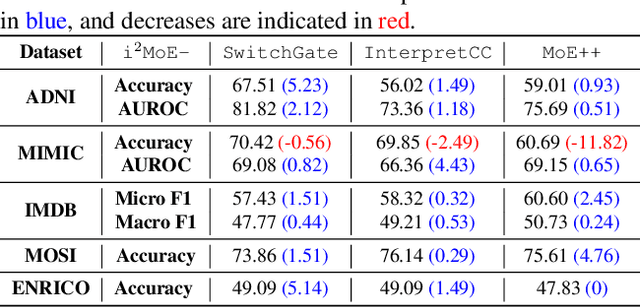
Modality fusion is a cornerstone of multimodal learning, enabling information integration from diverse data sources. However, vanilla fusion methods are limited by (1) inability to account for heterogeneous interactions between modalities and (2) lack of interpretability in uncovering the multimodal interactions inherent in the data. To this end, we propose I2MoE (Interpretable Multimodal Interaction-aware Mixture of Experts), an end-to-end MoE framework designed to enhance modality fusion by explicitly modeling diverse multimodal interactions, as well as providing interpretation on a local and global level. First, I2MoE utilizes different interaction experts with weakly supervised interaction losses to learn multimodal interactions in a data-driven way. Second, I2MoE deploys a reweighting model that assigns importance scores for the output of each interaction expert, which offers sample-level and dataset-level interpretation. Extensive evaluation of medical and general multimodal datasets shows that I2MoE is flexible enough to be combined with different fusion techniques, consistently improves task performance, and provides interpretation across various real-world scenarios. Code is available at https://github.com/Raina-Xin/I2MoE.
Flex-MoE: Modeling Arbitrary Modality Combination via the Flexible Mixture-of-Experts
Oct 10, 2024Multimodal learning has gained increasing importance across various fields, offering the ability to integrate data from diverse sources such as images, text, and personalized records, which are frequently observed in medical domains. However, in scenarios where some modalities are missing, many existing frameworks struggle to accommodate arbitrary modality combinations, often relying heavily on a single modality or complete data. This oversight of potential modality combinations limits their applicability in real-world situations. To address this challenge, we propose Flex-MoE (Flexible Mixture-of-Experts), a new framework designed to flexibly incorporate arbitrary modality combinations while maintaining robustness to missing data. The core idea of Flex-MoE is to first address missing modalities using a new missing modality bank that integrates observed modality combinations with the corresponding missing ones. This is followed by a uniquely designed Sparse MoE framework. Specifically, Flex-MoE first trains experts using samples with all modalities to inject generalized knowledge through the generalized router ($\mathcal{G}$-Router). The $\mathcal{S}$-Router then specializes in handling fewer modality combinations by assigning the top-1 gate to the expert corresponding to the observed modality combination. We evaluate Flex-MoE on the ADNI dataset, which encompasses four modalities in the Alzheimer's Disease domain, as well as on the MIMIC-IV dataset. The results demonstrate the effectiveness of Flex-MoE highlighting its ability to model arbitrary modality combinations in diverse missing modality scenarios. Code is available at https://github.com/UNITES-Lab/flex-moe.
Prefix-tree Decoding for Predicting Mass Spectra from Molecules
Mar 11, 2023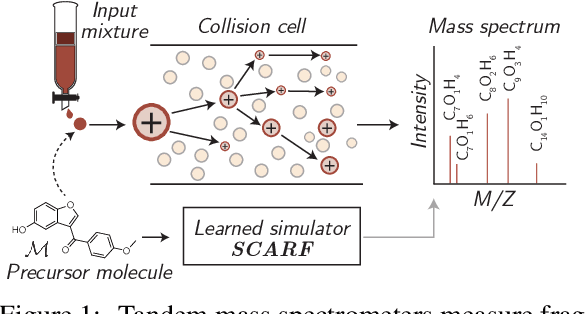
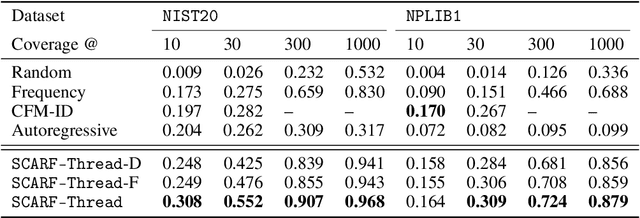
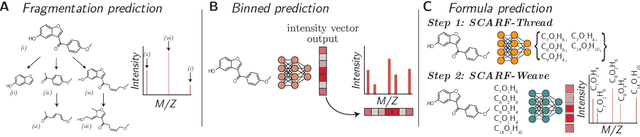
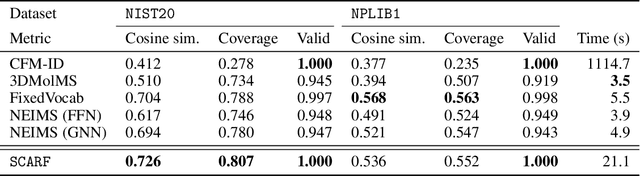
Computational predictions of mass spectra from molecules have enabled the discovery of clinically relevant metabolites. However, such predictive tools are still limited as they occupy one of two extremes, either operating (a) by fragmenting molecules combinatorially with overly rigid constraints on potential rearrangements and poor time complexity or (b) by decoding lossy and nonphysical discretized spectra vectors. In this work, we introduce a new intermediate strategy for predicting mass spectra from molecules by treating mass spectra as sets of chemical formulae, which are themselves multisets of atoms. After first encoding an input molecular graph, we decode a set of chemical subformulae, each of which specify a predicted peak in the mass spectra, the intensities of which are predicted by a second model. Our key insight is to overcome the combinatorial possibilities for chemical subformulae by decoding the formula set using a prefix tree structure, atom-type by atom-type, representing a general method for ordered multiset decoding. We show promising empirical results on mass spectra prediction tasks.
Retrieved Sequence Augmentation for Protein Representation Learning
Feb 24, 2023Protein language models have excelled in a variety of tasks, ranging from structure prediction to protein engineering. However, proteins are highly diverse in functions and structures, and current state-of-the-art models including the latest version of AlphaFold rely on Multiple Sequence Alignments (MSA) to feed in the evolutionary knowledge. Despite their success, heavy computational overheads, as well as the de novo and orphan proteins remain great challenges in protein representation learning. In this work, we show that MSAaugmented models inherently belong to retrievalaugmented methods. Motivated by this finding, we introduce Retrieved Sequence Augmentation(RSA) for protein representation learning without additional alignment or pre-processing. RSA links query protein sequences to a set of sequences with similar structures or properties in the database and combines these sequences for downstream prediction. We show that protein language models benefit from the retrieval enhancement on both structure prediction and property prediction tasks, with a 5% improvement on MSA Transformer on average while being 373 times faster. In addition, we show that our model can transfer to new protein domains better and outperforms MSA Transformer on de novo protein prediction. Our study fills a much-encountered gap in protein prediction and brings us a step closer to demystifying the domain knowledge needed to understand protein sequences. Code is available on https://github.com/HKUNLP/RSA.
Selective Annotation Makes Language Models Better Few-Shot Learners
Sep 05, 2022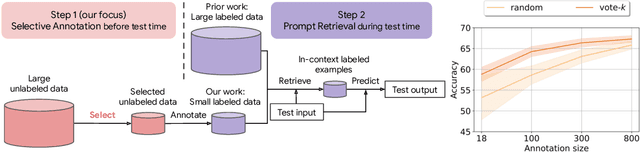
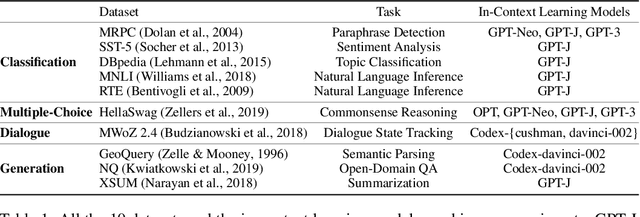
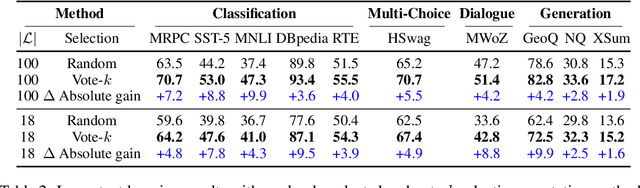
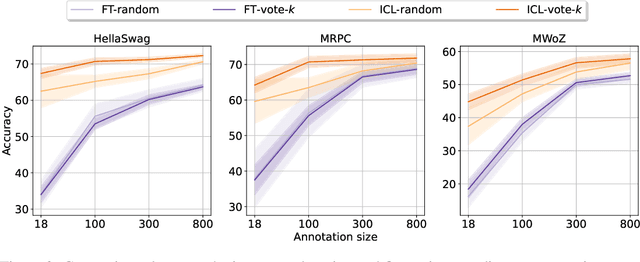
Many recent approaches to natural language tasks are built on the remarkable abilities of large language models. Large language models can perform in-context learning, where they learn a new task from a few task demonstrations, without any parameter updates. This work examines the implications of in-context learning for the creation of datasets for new natural language tasks. Departing from recent in-context learning methods, we formulate an annotation-efficient, two-step framework: selective annotation that chooses a pool of examples to annotate from unlabeled data in advance, followed by prompt retrieval that retrieves task examples from the annotated pool at test time. Based on this framework, we propose an unsupervised, graph-based selective annotation method, voke-k, to select diverse, representative examples to annotate. Extensive experiments on 10 datasets (covering classification, commonsense reasoning, dialogue, and text/code generation) demonstrate that our selective annotation method improves the task performance by a large margin. On average, vote-k achieves a 12.9%/11.4% relative gain under an annotation budget of 18/100, as compared to randomly selecting examples to annotate. Compared to state-of-the-art supervised finetuning approaches, it yields similar performance with 10-100x less annotation cost across 10 tasks. We further analyze the effectiveness of our framework in various scenarios: language models with varying sizes, alternative selective annotation methods, and cases where there is a test data domain shift. We hope that our studies will serve as a basis for data annotations as large language models are increasingly applied to new tasks. Our code is available at https://github.com/HKUNLP/icl-selective-annotation.