 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePushing the Boundaries of Natural Reasoning: Interleaved Bonus from Formal-Logic Verification
Jan 30, 2026Large Language Models (LLMs) show remarkable capabilities, yet their stochastic next-token prediction creates logical inconsistencies and reward hacking that formal symbolic systems avoid. To bridge this gap, we introduce a formal logic verification-guided framework that dynamically interleaves formal symbolic verification with the natural language generation process, providing real-time feedback to detect and rectify errors as they occur. Distinguished from previous neuro-symbolic methods limited by passive post-hoc validation, our approach actively penalizes intermediate fallacies during the reasoning chain. We operationalize this framework via a novel two-stage training pipeline that synergizes formal logic verification-guided supervised fine-tuning and policy optimization. Extensive evaluation on six benchmarks spanning mathematical, logical, and general reasoning demonstrates that our 7B and 14B models outperform state-of-the-art baselines by average margins of 10.4% and 14.2%, respectively. These results validate that formal verification can serve as a scalable mechanism to significantly push the performance boundaries of advanced LLM reasoning.
MMFineReason: Closing the Multimodal Reasoning Gap via Open Data-Centric Methods
Jan 29, 2026Recent advances in Vision Language Models (VLMs) have driven significant progress in visual reasoning. However, open-source VLMs still lag behind proprietary systems, largely due to the lack of high-quality reasoning data. Existing datasets offer limited coverage of challenging domains such as STEM diagrams and visual puzzles, and lack consistent, long-form Chain-of-Thought (CoT) annotations essential for eliciting strong reasoning capabilities. To bridge this gap, we introduce MMFineReason, a large-scale multimodal reasoning dataset comprising 1.8M samples and 5.1B solution tokens, featuring high-quality reasoning annotations distilled from Qwen3-VL-235B-A22B-Thinking. The dataset is established via a systematic three-stage pipeline: (1) large-scale data collection and standardization, (2) CoT rationale generation, and (3) comprehensive selection based on reasoning quality and difficulty awareness. The resulting dataset spans STEM problems, visual puzzles, games, and complex diagrams, with each sample annotated with visually grounded reasoning traces. We fine-tune Qwen3-VL-Instruct on MMFineReason to develop MMFineReason-2B/4B/8B versions. Our models establish new state-of-the-art results for their size class. Notably, MMFineReason-4B succesfully surpasses Qwen3-VL-8B-Thinking, and MMFineReason-8B even outperforms Qwen3-VL-30B-A3B-Thinking while approaching Qwen3-VL-32B-Thinking, demonstrating remarkable parameter efficiency. Crucially, we uncover a "less is more" phenomenon via our difficulty-aware filtering strategy: a subset of just 7\% (123K samples) achieves performance comparable to the full dataset. Notably, we reveal a synergistic effect where reasoning-oriented data composition simultaneously boosts general capabilities.
Parameter Inference and Uncertainty Quantification with Diffusion Models: Extending CDI to 2D Spatial Conditioning
Jan 23, 2026Uncertainty quantification is critical in scientific inverse problems to distinguish identifiable parameters from those that remain ambiguous given available measurements. The Conditional Diffusion Model-based Inverse Problem Solver (CDI) has previously demonstrated effective probabilistic inference for one-dimensional temporal signals, but its applicability to higher-dimensional spatial data remains unexplored. We extend CDI to two-dimensional spatial conditioning, enabling probabilistic parameter inference directly from spatial observations. We validate this extension on convergent beam electron diffraction (CBED) parameter inference - a challenging multi-parameter inverse problem in materials characterization where sample geometry, electronic structure, and thermal properties must be extracted from 2D diffraction patterns. Using simulated CBED data with ground-truth parameters, we demonstrate that CDI produces well-calibrated posterior distributions that accurately reflect measurement constraints: tight distributions for well-determined quantities and appropriately broad distributions for ambiguous parameters. In contrast, standard regression methods - while appearing accurate on aggregate metrics - mask this underlying uncertainty by predicting training set means for poorly constrained parameters. Our results confirm that CDI successfully extends from temporal to spatial domains, providing the genuine uncertainty information required for robust scientific inference.
ChartVerse: Scaling Chart Reasoning via Reliable Programmatic Synthesis from Scratch
Jan 20, 2026Chart reasoning is a critical capability for Vision Language Models (VLMs). However, the development of open-source models is severely hindered by the lack of high-quality training data. Existing datasets suffer from a dual challenge: synthetic charts are often simplistic and repetitive, while the associated QA pairs are prone to hallucinations and lack the reasoning depth required for complex tasks. To bridge this gap, we propose ChartVerse, a scalable framework designed to synthesize complex charts and reliable reasoning data from scratch. (1) To address the bottleneck of simple patterns, we first introduce Rollout Posterior Entropy (RPE), a novel metric that quantifies chart complexity. Guided by RPE, we develop complexity-aware chart coder to autonomously synthesize diverse, high-complexity charts via executable programs. (2) To guarantee reasoning rigor, we develop truth-anchored inverse QA synthesis. Diverging from standard generation, we adopt an answer-first paradigm: we extract deterministic answers directly from the source code, generate questions conditional on these anchors, and enforce strict consistency verification. To further elevate difficulty and reasoning depth, we filter samples based on model fail-rate and distill high-quality Chain-of-Thought (CoT) reasoning. We curate ChartVerse-SFT-600K and ChartVerse-RL-40K using Qwen3-VL-30B-A3B-Thinking as the teacher. Experimental results demonstrate that ChartVerse-8B achieves state-of-the-art performance, notably surpassing its teacher and rivaling the stronger Qwen3-VL-32B-Thinking.
Scientific Image Synthesis: Benchmarking, Methodologies, and Downstream Utility
Jan 17, 2026While synthetic data has proven effective for improving scientific reasoning in the text domain, multimodal reasoning remains constrained by the difficulty of synthesizing scientifically rigorous images. Existing Text-to-Image (T2I) models often produce outputs that are visually plausible yet scientifically incorrect, resulting in a persistent visual-logic divergence that limits their value for downstream reasoning. Motivated by recent advances in next-generation T2I models, we conduct a systematic study of scientific image synthesis across generation paradigms, evaluation, and downstream use. We analyze both direct pixel-based generation and programmatic synthesis, and propose ImgCoder, a logic-driven framework that follows an explicit "understand - plan - code" workflow to improve structural precision. To rigorously assess scientific correctness, we introduce SciGenBench, which evaluates generated images based on information utility and logical validity. Our evaluation reveals systematic failure modes in pixel-based models and highlights a fundamental expressiveness-precision trade-off. Finally, we show that fine-tuning Large Multimodal Models (LMMs) on rigorously verified synthetic scientific images yields consistent reasoning gains, with potential scaling trends analogous to the text domain, validating high-fidelity scientific synthesis as a viable path to unlocking massive multimodal reasoning capabilities.
LRAS: Advanced Legal Reasoning with Agentic Search
Jan 12, 2026While Large Reasoning Models (LRMs) have demonstrated exceptional logical capabilities in mathematical domains, their application to the legal field remains hindered by the strict requirements for procedural rigor and adherence to legal logic. Existing legal LLMs, which rely on "closed-loop reasoning" derived solely from internal parametric knowledge, frequently suffer from lack of self-awareness regarding their knowledge boundaries, leading to confident yet incorrect conclusions. To address this challenge, we present Legal Reasoning with Agentic Search (LRAS), the first framework designed to transition legal LLMs from static and parametric "closed-loop thinking" to dynamic and interactive "Active Inquiry". By integrating Introspective Imitation Learning and Difficulty-aware Reinforcement Learning, LRAS enables LRMs to identify knowledge boundaries and handle legal reasoning complexity. Empirical results demonstrate that LRAS outperforms state-of-the-art baselines by 8.2-32\%, with the most substantial gains observed in tasks requiring deep reasoning with reliable knowledge. We will release our data and models for further exploration soon.
SpatiaLoc: Leveraging Multi-Level Spatial Enhanced Descriptors for Cross-Modal Localization
Jan 07, 2026Cross-modal localization using text and point clouds enables robots to localize themselves via natural language descriptions, with applications in autonomous navigation and interaction between humans and robots. In this task, objects often recur across text and point clouds, making spatial relationships the most discriminative cues for localization. Given this characteristic, we present SpatiaLoc, a framework utilizing a coarse-to-fine strategy that emphasizes spatial relationships at both the instance and global levels. In the coarse stage, we introduce a Bezier Enhanced Object Spatial Encoder (BEOSE) that models spatial relationships at the instance level using quadratic Bezier curves. Additionally, a Frequency Aware Encoder (FAE) generates spatial representations in the frequency domain at the global level. In the fine stage, an Uncertainty Aware Gaussian Fine Localizer (UGFL) regresses 2D positions by modeling predictions as Gaussian distributions with a loss function aware of uncertainty. Extensive experiments on KITTI360Pose demonstrate that SpatiaLoc significantly outperforms existing state-of-the-art (SOTA) methods.
OpenDataArena: A Fair and Open Arena for Benchmarking Post-Training Dataset Value
Dec 16, 2025
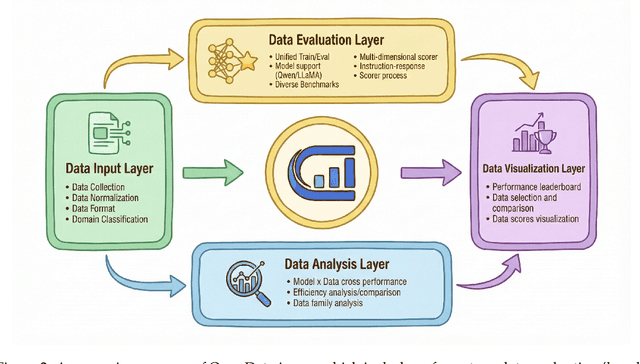

The rapid evolution of Large Language Models (LLMs) is predicated on the quality and diversity of post-training datasets. However, a critical dichotomy persists: while models are rigorously benchmarked, the data fueling them remains a black box--characterized by opaque composition, uncertain provenance, and a lack of systematic evaluation. This opacity hinders reproducibility and obscures the causal link between data characteristics and model behaviors. To bridge this gap, we introduce OpenDataArena (ODA), a holistic and open platform designed to benchmark the intrinsic value of post-training data. ODA establishes a comprehensive ecosystem comprising four key pillars: (i) a unified training-evaluation pipeline that ensures fair, open comparisons across diverse models (e.g., Llama, Qwen) and domains; (ii) a multi-dimensional scoring framework that profiles data quality along tens of distinct axes; (iii) an interactive data lineage explorer to visualize dataset genealogy and dissect component sources; and (iv) a fully open-source toolkit for training, evaluation, and scoring to foster data research. Extensive experiments on ODA--covering over 120 training datasets across multiple domains on 22 benchmarks, validated by more than 600 training runs and 40 million processed data points--reveal non-trivial insights. Our analysis uncovers the inherent trade-offs between data complexity and task performance, identifies redundancy in popular benchmarks through lineage tracing, and maps the genealogical relationships across datasets. We release all results, tools, and configurations to democratize access to high-quality data evaluation. Rather than merely expanding a leaderboard, ODA envisions a shift from trial-and-error data curation to a principled science of Data-Centric AI, paving the way for rigorous studies on data mixing laws and the strategic composition of foundation models.
GGBench: A Geometric Generative Reasoning Benchmark for Unified Multimodal Models
Nov 14, 2025
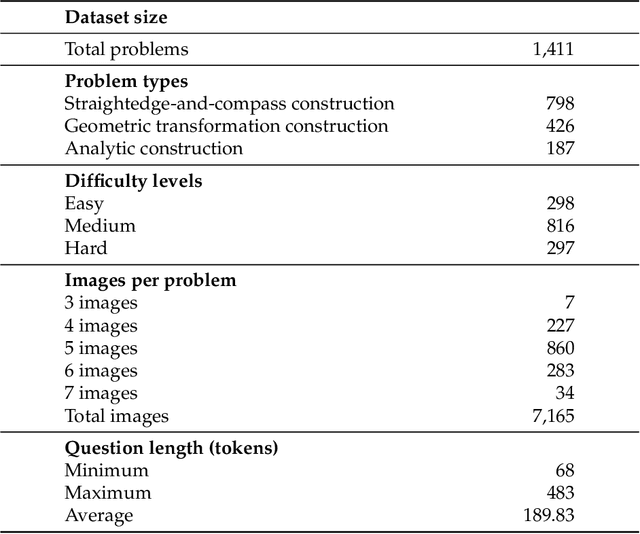
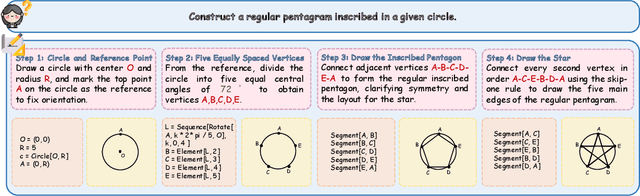

The advent of Unified Multimodal Models (UMMs) signals a paradigm shift in artificial intelligence, moving from passive perception to active, cross-modal generation. Despite their unprecedented ability to synthesize information, a critical gap persists in evaluation: existing benchmarks primarily assess discriminative understanding or unconstrained image generation separately, failing to measure the integrated cognitive process of generative reasoning. To bridge this gap, we propose that geometric construction provides an ideal testbed as it inherently demands a fusion of language comprehension and precise visual generation. We introduce GGBench, a benchmark designed specifically to evaluate geometric generative reasoning. It provides a comprehensive framework for systematically diagnosing a model's ability to not only understand and reason but to actively construct a solution, thereby setting a more rigorous standard for the next generation of intelligent systems. Project website: https://opendatalab-raiser.github.io/GGBench/.
Lost in Tokenization: Context as the Key to Unlocking Biomolecular Understanding in Scientific LLMs
Oct 27, 2025Scientific Large Language Models (Sci-LLMs) have emerged as a promising frontier for accelerating biological discovery. However, these models face a fundamental challenge when processing raw biomolecular sequences: the tokenization dilemma. Whether treating sequences as a specialized language, risking the loss of functional motif information, or as a separate modality, introducing formidable alignment challenges, current strategies fundamentally limit their reasoning capacity. We challenge this sequence-centric paradigm by positing that a more effective strategy is to provide Sci-LLMs with high-level structured context derived from established bioinformatics tools, thereby bypassing the need to interpret low-level noisy sequence data directly. Through a systematic comparison of leading Sci-LLMs on biological reasoning tasks, we tested three input modes: sequence-only, context-only, and a combination of both. Our findings are striking: the context-only approach consistently and substantially outperforms all other modes. Even more revealing, the inclusion of the raw sequence alongside its high-level context consistently degrades performance, indicating that raw sequences act as informational noise, even for models with specialized tokenization schemes. These results suggest that the primary strength of existing Sci-LLMs lies not in their nascent ability to interpret biomolecular syntax from scratch, but in their profound capacity for reasoning over structured, human-readable knowledge. Therefore, we argue for reframing Sci-LLMs not as sequence decoders, but as powerful reasoning engines over expert knowledge. This work lays the foundation for a new class of hybrid scientific AI agents, repositioning the developmental focus from direct sequence interpretation towards high-level knowledge synthesis. The code is available at github.com/opendatalab-raise-dev/CoKE.