 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTracing the Roots: A Multi-Agent Framework for Uncovering Data Lineage in Post-Training LLMs
Apr 12, 2026Post-training data plays a pivotal role in shaping the capabilities of Large Language Models (LLMs), yet datasets are often treated as isolated artifacts, overlooking the systemic connections that underlie their evolution. To disentangle these complex relationships, we introduce the concept of \textbf{data lineage} to the LLM ecosystem and propose an automated multi-agent framework to reconstruct the evolutionary graph of dataset development. Through large-scale lineage analysis, we characterize domain-specific structural patterns, such as vertical refinement in math-oriented datasets and horizontal aggregation in general-domain corpora. Moreover, we uncover pervasive systemic issues, including \textit{structural redundancy} induced by implicit dataset intersections and the \textit{propagation of benchmark contamination} along lineage paths. To demonstrate the practical value of lineage analysis for data construction, we leverage the reconstructed lineage graph to create a \textit{lineage-aware diversity-oriented dataset}. By anchoring instruction sampling at upstream root sources, this approach mitigates downstream homogenization and hidden redundancy, yielding a more diverse post-training corpus. We further highlight lineage-centric analysis as an efficient and robust topological alternative to sample-level dataset comparison for large-scale data ecosystems. By grounding data construction in explicit lineage structures, our work advances post-training data curation toward a more systematic and controllable paradigm.
Intern-S1-Pro: Scientific Multimodal Foundation Model at Trillion Scale
Mar 26, 2026We introduce Intern-S1-Pro, the first one-trillion-parameter scientific multimodal foundation model. Scaling to this unprecedented size, the model delivers a comprehensive enhancement across both general and scientific domains. Beyond stronger reasoning and image-text understanding capabilities, its intelligence is augmented with advanced agent capabilities. Simultaneously, its scientific expertise has been vastly expanded to master over 100 specialized tasks across critical science fields, including chemistry, materials, life sciences, and earth sciences. Achieving this massive scale is made possible by the robust infrastructure support of XTuner and LMDeploy, which facilitates highly efficient Reinforcement Learning (RL) training at the 1-trillion parameter level while ensuring strict precision consistency between training and inference. By seamlessly integrating these advancements, Intern-S1-Pro further fortifies the fusion of general and specialized intelligence, working as a Specializable Generalist, demonstrating its position in the top tier of open-source models for general capabilities, while outperforming proprietary models in the depth of specialized scientific tasks.
ChartVerse: Scaling Chart Reasoning via Reliable Programmatic Synthesis from Scratch
Jan 20, 2026Chart reasoning is a critical capability for Vision Language Models (VLMs). However, the development of open-source models is severely hindered by the lack of high-quality training data. Existing datasets suffer from a dual challenge: synthetic charts are often simplistic and repetitive, while the associated QA pairs are prone to hallucinations and lack the reasoning depth required for complex tasks. To bridge this gap, we propose ChartVerse, a scalable framework designed to synthesize complex charts and reliable reasoning data from scratch. (1) To address the bottleneck of simple patterns, we first introduce Rollout Posterior Entropy (RPE), a novel metric that quantifies chart complexity. Guided by RPE, we develop complexity-aware chart coder to autonomously synthesize diverse, high-complexity charts via executable programs. (2) To guarantee reasoning rigor, we develop truth-anchored inverse QA synthesis. Diverging from standard generation, we adopt an answer-first paradigm: we extract deterministic answers directly from the source code, generate questions conditional on these anchors, and enforce strict consistency verification. To further elevate difficulty and reasoning depth, we filter samples based on model fail-rate and distill high-quality Chain-of-Thought (CoT) reasoning. We curate ChartVerse-SFT-600K and ChartVerse-RL-40K using Qwen3-VL-30B-A3B-Thinking as the teacher. Experimental results demonstrate that ChartVerse-8B achieves state-of-the-art performance, notably surpassing its teacher and rivaling the stronger Qwen3-VL-32B-Thinking.
Scientific Image Synthesis: Benchmarking, Methodologies, and Downstream Utility
Jan 17, 2026While synthetic data has proven effective for improving scientific reasoning in the text domain, multimodal reasoning remains constrained by the difficulty of synthesizing scientifically rigorous images. Existing Text-to-Image (T2I) models often produce outputs that are visually plausible yet scientifically incorrect, resulting in a persistent visual-logic divergence that limits their value for downstream reasoning. Motivated by recent advances in next-generation T2I models, we conduct a systematic study of scientific image synthesis across generation paradigms, evaluation, and downstream use. We analyze both direct pixel-based generation and programmatic synthesis, and propose ImgCoder, a logic-driven framework that follows an explicit "understand - plan - code" workflow to improve structural precision. To rigorously assess scientific correctness, we introduce SciGenBench, which evaluates generated images based on information utility and logical validity. Our evaluation reveals systematic failure modes in pixel-based models and highlights a fundamental expressiveness-precision trade-off. Finally, we show that fine-tuning Large Multimodal Models (LMMs) on rigorously verified synthetic scientific images yields consistent reasoning gains, with potential scaling trends analogous to the text domain, validating high-fidelity scientific synthesis as a viable path to unlocking massive multimodal reasoning capabilities.
OpenDataArena: A Fair and Open Arena for Benchmarking Post-Training Dataset Value
Dec 16, 2025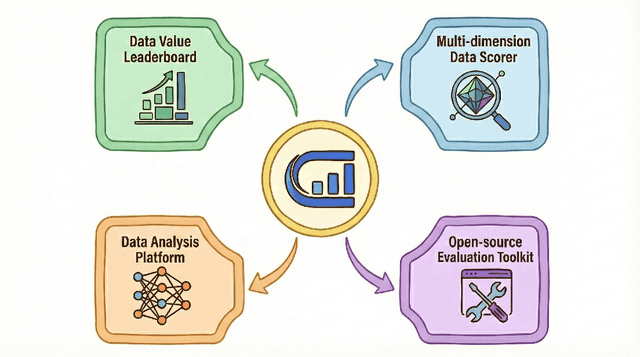
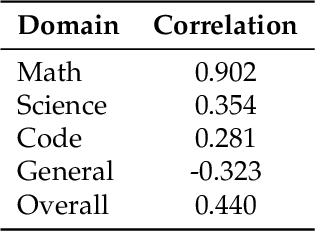
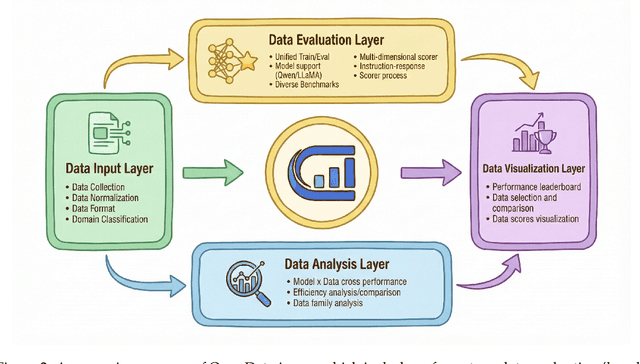

The rapid evolution of Large Language Models (LLMs) is predicated on the quality and diversity of post-training datasets. However, a critical dichotomy persists: while models are rigorously benchmarked, the data fueling them remains a black box--characterized by opaque composition, uncertain provenance, and a lack of systematic evaluation. This opacity hinders reproducibility and obscures the causal link between data characteristics and model behaviors. To bridge this gap, we introduce OpenDataArena (ODA), a holistic and open platform designed to benchmark the intrinsic value of post-training data. ODA establishes a comprehensive ecosystem comprising four key pillars: (i) a unified training-evaluation pipeline that ensures fair, open comparisons across diverse models (e.g., Llama, Qwen) and domains; (ii) a multi-dimensional scoring framework that profiles data quality along tens of distinct axes; (iii) an interactive data lineage explorer to visualize dataset genealogy and dissect component sources; and (iv) a fully open-source toolkit for training, evaluation, and scoring to foster data research. Extensive experiments on ODA--covering over 120 training datasets across multiple domains on 22 benchmarks, validated by more than 600 training runs and 40 million processed data points--reveal non-trivial insights. Our analysis uncovers the inherent trade-offs between data complexity and task performance, identifies redundancy in popular benchmarks through lineage tracing, and maps the genealogical relationships across datasets. We release all results, tools, and configurations to democratize access to high-quality data evaluation. Rather than merely expanding a leaderboard, ODA envisions a shift from trial-and-error data curation to a principled science of Data-Centric AI, paving the way for rigorous studies on data mixing laws and the strategic composition of foundation models.
Middo: Model-Informed Dynamic Data Optimization for Enhanced LLM Fine-Tuning via Closed-Loop Learning
Aug 29, 2025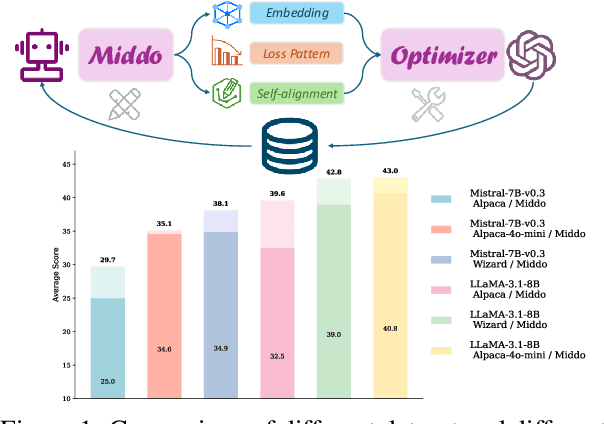
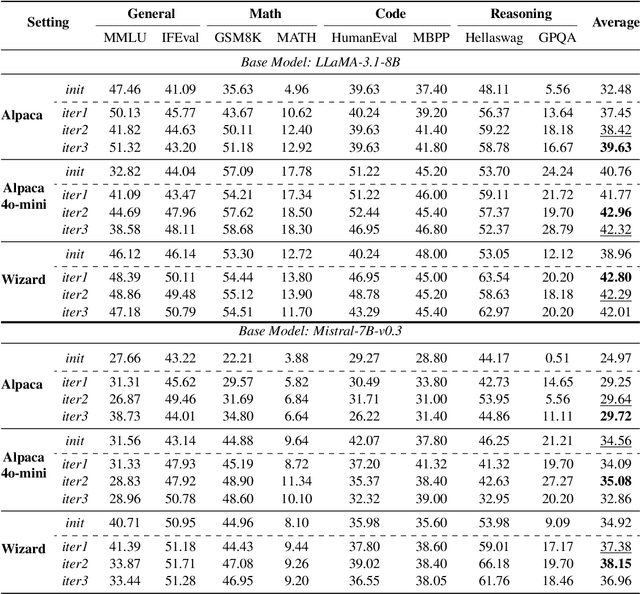
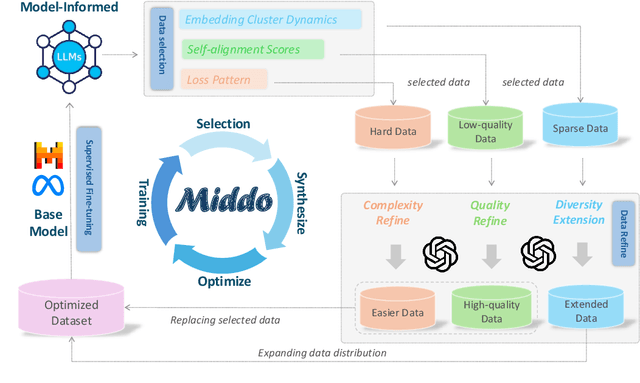
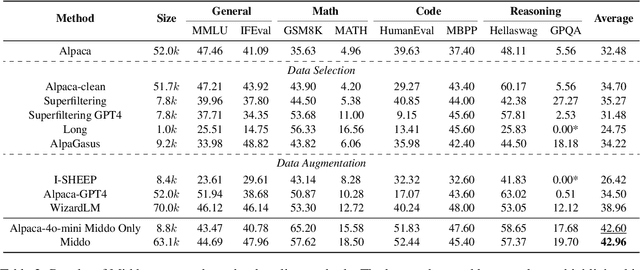
Supervised Fine-Tuning (SFT) Large Language Models (LLM) fundamentally rely on high-quality training data. While data selection and data synthesis are two common strategies to improve data quality, existing approaches often face limitations in static dataset curation that fail to adapt to evolving model capabilities. In this paper, we introduce Middo, a self-evolving Model-informed dynamic data optimization framework that uses model-aware data selection and context-preserving data refinement. Unlike conventional one-off filtering/synthesis methods, our framework establishes a closed-loop optimization system: (1) A self-referential diagnostic module proactively identifies suboptimal samples through tri-axial model signals - loss patterns (complexity), embedding cluster dynamics (diversity), and self-alignment scores (quality); (2) An adaptive optimization engine then transforms suboptimal samples into pedagogically valuable training points while preserving semantic integrity; (3) This optimization process continuously evolves with model capability through dynamic learning principles. Experiments on multiple benchmarks demonstrate that our \method consistently enhances the quality of seed data and boosts LLM's performance with improving accuracy by 7.15% on average while maintaining the original dataset scale. This work establishes a new paradigm for sustainable LLM training through dynamic human-AI co-evolution of data and models. Our datasets, models, and code are coming soon.
IDEAL: Data Equilibrium Adaptation for Multi-Capability Language Model Alignment
May 19, 2025



Large Language Models (LLMs) have achieved impressive performance through Supervised Fine-tuning (SFT) on diverse instructional datasets. When training on multiple capabilities simultaneously, the mixture training dataset, governed by volumes of data from different domains, is a critical factor that directly impacts the final model's performance. Unlike many studies that focus on enhancing the quality of training datasets through data selection methods, few works explore the intricate relationship between the compositional quantity of mixture training datasets and the emergent capabilities of LLMs. Given the availability of a high-quality multi-domain training dataset, understanding the impact of data from each domain on the model's overall capabilities is crucial for preparing SFT data and training a well-balanced model that performs effectively across diverse domains. In this work, we introduce IDEAL, an innovative data equilibrium adaptation framework designed to effectively optimize volumes of data from different domains within mixture SFT datasets, thereby enhancing the model's alignment and performance across multiple capabilities. IDEAL employs a gradient-based approach to iteratively refine the training data distribution, dynamically adjusting the volumes of domain-specific data based on their impact on downstream task performance. By leveraging this adaptive mechanism, IDEAL ensures a balanced dataset composition, enabling the model to achieve robust generalization and consistent proficiency across diverse tasks. Experiments across different capabilities demonstrate that IDEAL outperforms conventional uniform data allocation strategies, achieving a comprehensive improvement of approximately 7% in multi-task evaluation scores.
CipherBank: Exploring the Boundary of LLM Reasoning Capabilities through Cryptography Challenges
Apr 27, 2025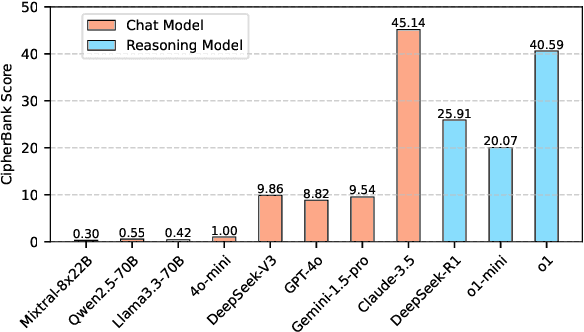



Large language models (LLMs) have demonstrated remarkable capabilities, especially the recent advancements in reasoning, such as o1 and o3, pushing the boundaries of AI. Despite these impressive achievements in mathematics and coding, the reasoning abilities of LLMs in domains requiring cryptographic expertise remain underexplored. In this paper, we introduce CipherBank, a comprehensive benchmark designed to evaluate the reasoning capabilities of LLMs in cryptographic decryption tasks. CipherBank comprises 2,358 meticulously crafted problems, covering 262 unique plaintexts across 5 domains and 14 subdomains, with a focus on privacy-sensitive and real-world scenarios that necessitate encryption. From a cryptographic perspective, CipherBank incorporates 3 major categories of encryption methods, spanning 9 distinct algorithms, ranging from classical ciphers to custom cryptographic techniques. We evaluate state-of-the-art LLMs on CipherBank, e.g., GPT-4o, DeepSeek-V3, and cutting-edge reasoning-focused models such as o1 and DeepSeek-R1. Our results reveal significant gaps in reasoning abilities not only between general-purpose chat LLMs and reasoning-focused LLMs but also in the performance of current reasoning-focused models when applied to classical cryptographic decryption tasks, highlighting the challenges these models face in understanding and manipulating encrypted data. Through detailed analysis and error investigations, we provide several key observations that shed light on the limitations and potential improvement areas for LLMs in cryptographic reasoning. These findings underscore the need for continuous advancements in LLM reasoning capabilities.
MathFusion: Enhancing Mathematic Problem-solving of LLM through Instruction Fusion
Mar 20, 2025Large Language Models (LLMs) have shown impressive progress in mathematical reasoning. While data augmentation is promising to enhance mathematical problem-solving ability, current approaches are predominantly limited to instance-level modifications-such as rephrasing or generating syntactic variations-which fail to capture and leverage the intrinsic relational structures inherent in mathematical knowledge. Inspired by human learning processes, where mathematical proficiency develops through systematic exposure to interconnected concepts, we introduce MathFusion, a novel framework that enhances mathematical reasoning through cross-problem instruction synthesis. MathFusion implements this through three fusion strategies: (1) sequential fusion, which chains related problems to model solution dependencies; (2) parallel fusion, which combines analogous problems to reinforce conceptual understanding; and (3) conditional fusion, which creates context-aware selective problems to enhance reasoning flexibility. By applying these strategies, we generate a new dataset, \textbf{MathFusionQA}, followed by fine-tuning models (DeepSeekMath-7B, Mistral-7B, Llama3-8B) on it. Experimental results demonstrate that MathFusion achieves substantial improvements in mathematical reasoning while maintaining high data efficiency, boosting performance by 18.0 points in accuracy across diverse benchmarks while requiring only 45K additional synthetic instructions, representing a substantial improvement over traditional single-instruction approaches. Our datasets, models, and code are publicly available at https://github.com/QizhiPei/mathfusion.
MetaLadder: Ascending Mathematical Solution Quality via Analogical-Problem Reasoning Transfer
Mar 19, 2025Large Language Models (LLMs) have demonstrated promising capabilities in solving mathematical reasoning tasks, leveraging Chain-of-Thought (CoT) data as a vital component in guiding answer generation. Current paradigms typically generate CoT and answers directly for a given problem, diverging from human problem-solving strategies to some extent. Humans often solve problems by recalling analogous cases and leveraging their solutions to reason about the current task. Inspired by this cognitive process, we propose \textbf{MetaLadder}, a novel framework that explicitly prompts LLMs to recall and reflect on meta-problems, those structurally or semantically analogous problems, alongside their CoT solutions before addressing the target problem. Additionally, we introduce a problem-restating mechanism to enhance the model's comprehension of the target problem by regenerating the original question, which further improves reasoning accuracy. Therefore, the model can achieve reasoning transfer from analogical problems, mimicking human-like "learning from examples" and generalization abilities. Extensive experiments on mathematical benchmarks demonstrate that our MetaLadder significantly boosts LLMs' problem-solving accuracy, largely outperforming standard CoT-based methods (\textbf{10.3\%} accuracy gain) and other methods. Our code and data has been released at https://github.com/LHL3341/MetaLadder.