 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-Level Aware Preference Learning: Enhancing RLHF for Complex Multi-Instruction Tasks
May 19, 2025



RLHF has emerged as a predominant approach for aligning artificial intelligence systems with human preferences, demonstrating exceptional and measurable efficacy in instruction following tasks; however, it exhibits insufficient compliance capabilities when confronted with complex multi-instruction tasks. Conventional approaches rely heavily on human annotation or more sophisticated large language models, thereby introducing substantial resource expenditure or potential bias concerns. Meanwhile, alternative synthetic methods that augment standard preference datasets often compromise the model's semantic quality. Our research identifies a critical oversight in existing techniques, which predominantly focus on comparing responses while neglecting valuable latent signals embedded within prompt inputs, and which only focus on preference disparities at the intra-sample level, while neglecting to account for the inter-sample level preference differentials that exist among preference data. To leverage these previously neglected indicators, we propose a novel Multi-level Aware Preference Learning (MAPL) framework, capable of enhancing multi-instruction capabilities. Specifically, for any given response in original preference data pairs, we construct varied prompts with a preference relation under different conditions, in order to learn intra-sample level preference disparities. Furthermore, for any given original preference pair, we synthesize multi-instruction preference pairs to capture preference discrepancies at the inter-sample level. Building on the two datasets constructed above, we consequently devise two sophisticated training objective functions. Subsequently, our framework integrates seamlessly into both Reward Modeling and Direct Preference Optimization paradigms. Through rigorous evaluation across multiple benchmarks, we empirically validate the efficacy of our framework.
Bias Fitting to Mitigate Length Bias of Reward Model in RLHF
May 19, 2025



Reinforcement Learning from Human Feedback relies on reward models to align large language models with human preferences. However, RLHF often suffers from reward hacking, wherein policy learning exploits flaws in the trained reward model to maximize reward scores without genuinely aligning with human preferences. A significant example of such reward hacking is length bias, where reward models usually favor longer responses irrespective of actual response quality. Previous works on length bias have notable limitations, these approaches either mitigate bias without characterizing the bias form, or simply assume a linear length-reward relation. To accurately model the intricate nature of length bias and facilitate more effective bias mitigation, we propose FiMi-RM (Bias Fitting to Mitigate Length Bias of Reward Model in RLHF), a framework that autonomously learns and corrects underlying bias patterns. Our approach consists of three stages: First, we train a standard reward model which inherently contains length bias. Next, we deploy a lightweight fitting model to explicitly capture the non-linear relation between length and reward. Finally, we incorporate this learned relation into the reward model to debias. Experimental results demonstrate that FiMi-RM achieves a more balanced length-reward distribution. Furthermore, when applied to alignment algorithms, our debiased reward model improves length-controlled win rate and reduces verbosity without compromising its performance.
DAPO: An Open-Source LLM Reinforcement Learning System at Scale
Mar 18, 2025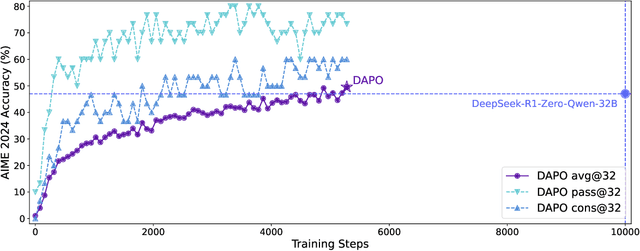

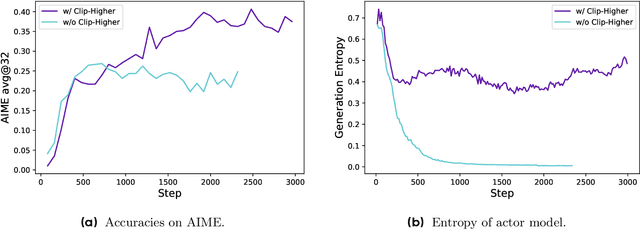

Inference scaling empowers LLMs with unprecedented reasoning ability, with reinforcement learning as the core technique to elicit complex reasoning. However, key technical details of state-of-the-art reasoning LLMs are concealed (such as in OpenAI o1 blog and DeepSeek R1 technical report), thus the community still struggles to reproduce their RL training results. We propose the $\textbf{D}$ecoupled Clip and $\textbf{D}$ynamic s$\textbf{A}$mpling $\textbf{P}$olicy $\textbf{O}$ptimization ($\textbf{DAPO}$) algorithm, and fully open-source a state-of-the-art large-scale RL system that achieves 50 points on AIME 2024 using Qwen2.5-32B base model. Unlike previous works that withhold training details, we introduce four key techniques of our algorithm that make large-scale LLM RL a success. In addition, we open-source our training code, which is built on the verl framework, along with a carefully curated and processed dataset. These components of our open-source system enhance reproducibility and support future research in large-scale LLM RL.
Reveal the Mystery of DPO: The Connection between DPO and RL Algorithms
Feb 05, 2025With the rapid development of Large Language Models (LLMs), numerous Reinforcement Learning from Human Feedback (RLHF) algorithms have been introduced to improve model safety and alignment with human preferences. These algorithms can be divided into two main frameworks based on whether they require an explicit reward (or value) function for training: actor-critic-based Proximal Policy Optimization (PPO) and alignment-based Direct Preference Optimization (DPO). The mismatch between DPO and PPO, such as DPO's use of a classification loss driven by human-preferred data, has raised confusion about whether DPO should be classified as a Reinforcement Learning (RL) algorithm. To address these ambiguities, we focus on three key aspects related to DPO, RL, and other RLHF algorithms: (1) the construction of the loss function; (2) the target distribution at which the algorithm converges; (3) the impact of key components within the loss function. Specifically, we first establish a unified framework named UDRRA connecting these algorithms based on the construction of their loss functions. Next, we uncover their target policy distributions within this framework. Finally, we investigate the critical components of DPO to understand their impact on the convergence rate. Our work provides a deeper understanding of the relationship between DPO, RL, and other RLHF algorithms, offering new insights for improving existing algorithms.
Disentangling Length Bias In Preference Learning Via Response-Conditioned Modeling
Feb 02, 2025



Reinforcement Learning from Human Feedback (RLHF) has achieved considerable success in aligning large language models (LLMs) by modeling human preferences with a learnable reward model and employing a reinforcement learning algorithm to maximize the reward model's scores. However, these reward models are susceptible to exploitation through various superficial confounding factors, with length bias emerging as a particularly significant concern. Moreover, while the pronounced impact of length bias on preference modeling suggests that LLMs possess an inherent sensitivity to length perception, our preliminary investigations reveal that fine-tuned LLMs consistently struggle to adhere to explicit length instructions. To address these two limitations, we propose a novel framework wherein the reward model explicitly differentiates between human semantic preferences and response length requirements. Specifically, we introduce a Response-conditioned Bradley-Terry (Rc-BT) model that enhances the reward model's capability in length bias mitigating and length instruction following, through training on our augmented dataset. Furthermore, we propose the Rc-DPO algorithm to leverage the Rc-BT model for direct policy optimization (DPO) of LLMs, simultaneously mitigating length bias and promoting adherence to length instructions. Extensive evaluations demonstrate that our approach substantially improves both preference modeling and length instruction compliance, with its effectiveness validated across various foundational models and preference datasets.
BoolQuestions: Does Dense Retrieval Understand Boolean Logic in Language?
Nov 19, 2024



Dense retrieval, which aims to encode the semantic information of arbitrary text into dense vector representations or embeddings, has emerged as an effective and efficient paradigm for text retrieval, consequently becoming an essential component in various natural language processing systems. These systems typically focus on optimizing the embedding space by attending to the relevance of text pairs, while overlooking the Boolean logic inherent in language, which may not be captured by current training objectives. In this work, we first investigate whether current retrieval systems can comprehend the Boolean logic implied in language. To answer this question, we formulate the task of Boolean Dense Retrieval and collect a benchmark dataset, BoolQuestions, which covers complex queries containing basic Boolean logic and corresponding annotated passages. Through extensive experimental results on the proposed task and benchmark dataset, we draw the conclusion that current dense retrieval systems do not fully understand Boolean logic in language, and there is a long way to go to improve our dense retrieval systems. Furthermore, to promote further research on enhancing the understanding of Boolean logic for language models, we explore Boolean operation on decomposed query and propose a contrastive continual training method that serves as a strong baseline for the research community.
* Findings of the Association for Computational Linguistics: EMNLP 2024
Trustworthy Alignment of Retrieval-Augmented Large Language Models via Reinforcement Learning
Oct 22, 2024



Trustworthiness is an essential prerequisite for the real-world application of large language models. In this paper, we focus on the trustworthiness of language models with respect to retrieval augmentation. Despite being supported with external evidence, retrieval-augmented generation still suffers from hallucinations, one primary cause of which is the conflict between contextual and parametric knowledge. We deem that retrieval-augmented language models have the inherent capabilities of supplying response according to both contextual and parametric knowledge. Inspired by aligning language models with human preference, we take the first step towards aligning retrieval-augmented language models to a status where it responds relying merely on the external evidence and disregards the interference of parametric knowledge. Specifically, we propose a reinforcement learning based algorithm Trustworthy-Alignment, theoretically and experimentally demonstrating large language models' capability of reaching a trustworthy status without explicit supervision on how to respond. Our work highlights the potential of large language models on exploring its intrinsic abilities by its own and expands the application scenarios of alignment from fulfilling human preference to creating trustworthy agents.
* ICML 2024
Exploiting Pre-trained Models for Drug Target Affinity Prediction with Nearest Neighbors
Jul 21, 2024



Drug-Target binding Affinity (DTA) prediction is essential for drug discovery. Despite the application of deep learning methods to DTA prediction, the achieved accuracy remain suboptimal. In this work, inspired by the recent success of retrieval methods, we propose $k$NN-DTA, a non-parametric embedding-based retrieval method adopted on a pre-trained DTA prediction model, which can extend the power of the DTA model with no or negligible cost. Different from existing methods, we introduce two neighbor aggregation ways from both embedding space and label space that are integrated into a unified framework. Specifically, we propose a \emph{label aggregation} with \emph{pair-wise retrieval} and a \emph{representation aggregation} with \emph{point-wise retrieval} of the nearest neighbors. This method executes in the inference phase and can efficiently boost the DTA prediction performance with no training cost. In addition, we propose an extension, Ada-$k$NN-DTA, an instance-wise and adaptive aggregation with lightweight learning. Results on four benchmark datasets show that $k$NN-DTA brings significant improvements, outperforming previous state-of-the-art (SOTA) results, e.g, on BindingDB IC$_{50}$ and $K_i$ testbeds, $k$NN-DTA obtains new records of RMSE $\bf{0.684}$ and $\bf{0.750}$. The extended Ada-$k$NN-DTA further improves the performance to be $\bf{0.675}$ and $\bf{0.735}$ RMSE. These results strongly prove the effectiveness of our method. Results in other settings and comprehensive studies/analyses also show the great potential of our $k$NN-DTA approach.
3D-MolT5: Towards Unified 3D Molecule-Text Modeling with 3D Molecular Tokenization
Jun 09, 2024



The integration of molecule and language has garnered increasing attention in molecular science. Recent advancements in Language Models (LMs) have demonstrated potential for the comprehensive modeling of molecule and language. However, existing works exhibit notable limitations. Most existing works overlook the modeling of 3D information, which is crucial for understanding molecular structures and also functions. While some attempts have been made to leverage external structure encoding modules to inject the 3D molecular information into LMs, there exist obvious difficulties that hinder the integration of molecular structure and language text, such as modality alignment and separate tuning. To bridge this gap, we propose 3D-MolT5, a unified framework designed to model both 1D molecular sequence and 3D molecular structure. The key innovation lies in our methodology for mapping fine-grained 3D substructure representations (based on 3D molecular fingerprints) to a specialized 3D token vocabulary for 3D-MolT5. This 3D structure token vocabulary enables the seamless combination of 1D sequence and 3D structure representations in a tokenized format, allowing 3D-MolT5 to encode molecular sequence (SELFIES), molecular structure, and text sequences within a unified architecture. Alongside, we further introduce 1D and 3D joint pre-training to enhance the model's comprehension of these diverse modalities in a joint representation space and better generalize to various tasks for our foundation model. Through instruction tuning on multiple downstream datasets, our proposed 3D-MolT5 shows superior performance than existing methods in molecular property prediction, molecule captioning, and text-based molecule generation tasks. Our code will be available on GitHub soon.
FABind+: Enhancing Molecular Docking through Improved Pocket Prediction and Pose Generation
Apr 07, 2024



Molecular docking is a pivotal process in drug discovery. While traditional techniques rely on extensive sampling and simulation governed by physical principles, these methods are often slow and costly. The advent of deep learning-based approaches has shown significant promise, offering increases in both accuracy and efficiency. Building upon the foundational work of FABind, a model designed with a focus on speed and accuracy, we present FABind+, an enhanced iteration that largely boosts the performance of its predecessor. We identify pocket prediction as a critical bottleneck in molecular docking and propose a novel methodology that significantly refines pocket prediction, thereby streamlining the docking process. Furthermore, we introduce modifications to the docking module to enhance its pose generation capabilities. In an effort to bridge the gap with conventional sampling/generative methods, we incorporate a simple yet effective sampling technique coupled with a confidence model, requiring only minor adjustments to the regression framework of FABind. Experimental results and analysis reveal that FABind+ remarkably outperforms the original FABind, achieves competitive state-of-the-art performance, and delivers insightful modeling strategies. This demonstrates FABind+ represents a substantial step forward in molecular docking and drug discovery. Our code is in https://github.com/QizhiPei/FABind.