 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSAFE-QAQ: End-to-End Slow-Thinking Audio-Text Fraud Detection via Reinforcement Learning
Jan 04, 2026Existing fraud detection methods predominantly rely on transcribed text, suffering from ASR errors and missing crucial acoustic cues like vocal tone and environmental context. This limits their effectiveness against complex deceptive strategies. To address these challenges, we first propose \textbf{SAFE-QAQ}, an end-to-end comprehensive framework for audio-based slow-thinking fraud detection. First, the SAFE-QAQ framework eliminates the impact of transcription errors on detection performance. Secondly, we propose rule-based slow-thinking reward mechanisms that systematically guide the system to identify fraud-indicative patterns by accurately capturing fine-grained audio details, through hierarchical reasoning processes. Besides, our framework introduces a dynamic risk assessment framework during live calls, enabling early detection and prevention of fraud. Experiments on the TeleAntiFraud-Bench demonstrate that SAFE-QAQ achieves dramatic improvements over existing methods in multiple key dimensions, including accuracy, inference efficiency, and real-time processing capabilities. Currently deployed and analyzing over 70,000 calls daily, SAFE-QAQ effectively automates complex fraud detection, reducing human workload and financial losses. Code: https://anonymous.4open.science/r/SAFE-QAQ.
HI-TransPA: Hearing Impairments Translation Personal Assistant
Nov 14, 2025Hearing-impaired individuals often face significant barriers in daily communication due to the inherent challenges of producing clear speech. To address this, we introduce the Omni-Model paradigm into assistive technology and present HI-TransPA, an instruction-driven audio-visual personal assistant. The model fuses indistinct speech with lip dynamics, enabling both translation and dialogue within a single multimodal framework. To address the distinctive pronunciation patterns of hearing-impaired speech and the limited adaptability of existing models, we develop a multimodal preprocessing and curation pipeline that detects facial landmarks, stabilizes the lip region, and quantitatively evaluates sample quality. These quality scores guide a curriculum learning strategy that first trains on clean, high-confidence samples and progressively incorporates harder cases to strengthen model robustness. Architecturally, we employs a novel unified 3D-Resampler to efficiently encode the lip dynamics, which is critical for accurate interpretation. Experiments on purpose-built HI-Dialogue dataset show that HI-TransPA achieves state-of-the-art performance in both literal accuracy and semantic fidelity. Our work establishes a foundation for applying Omni-Models to assistive communication technology, providing an end-to-end modeling framework and essential processing tools for future research.
DGRO: Enhancing LLM Reasoning via Exploration-Exploitation Control and Reward Variance Management
May 19, 2025
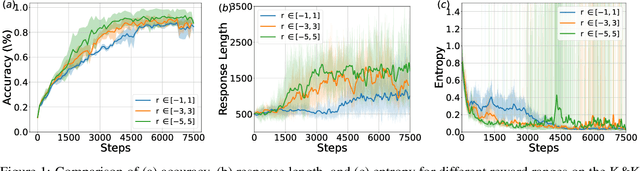
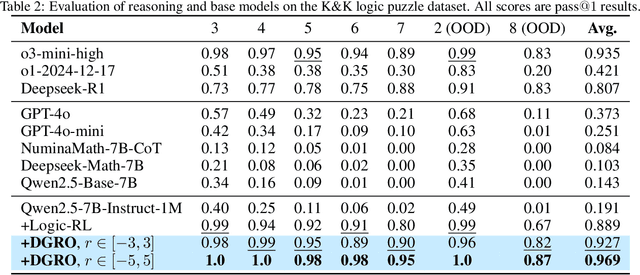
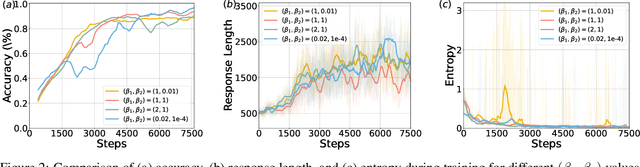
Inference scaling further accelerates Large Language Models (LLMs) toward Artificial General Intelligence (AGI), with large-scale Reinforcement Learning (RL) to unleash long Chain-of-Thought reasoning. Most contemporary reasoning approaches usually rely on handcrafted rule-based reward functions. However, the tarde-offs of exploration and exploitation in RL algorithms involves multiple complex considerations, and the theoretical and empirical impacts of manually designed reward functions remain insufficiently explored. In this paper, we propose Decoupled Group Reward Optimization (DGRO), a general RL algorithm for LLM reasoning. On the one hand, DGRO decouples the traditional regularization coefficient into two independent hyperparameters: one scales the policy gradient term, and the other regulates the distance from the sampling policy. This decoupling not only enables precise control over balancing exploration and exploitation, but also can be seamlessly extended to Online Policy Mirror Descent (OPMD) algorithms in Kimi k1.5 and Direct Reward Optimization. On the other hand, we observe that reward variance significantly affects both convergence speed and final model performance. We conduct both theoretical analysis and extensive empirical validation to assess DGRO, including a detailed ablation study that investigates its performance and optimization dynamics. Experimental results show that DGRO achieves state-of-the-art performance on the Logic dataset with an average accuracy of 96.9\%, and demonstrates strong generalization across mathematical benchmarks.
Trust Region Preference Approximation: A simple and stable reinforcement learning algorithm for LLM reasoning
Apr 06, 2025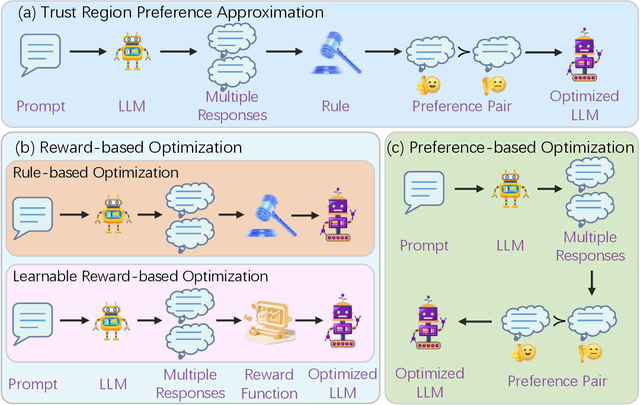
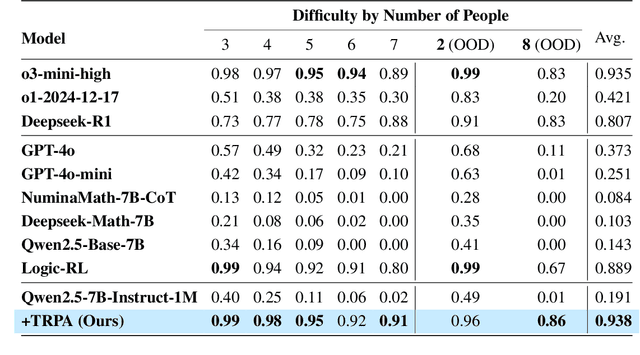
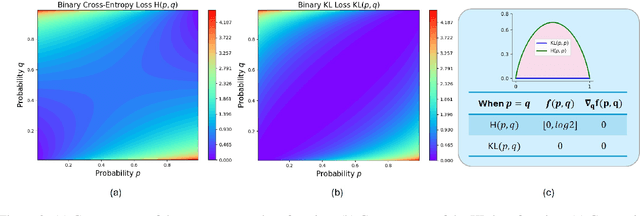
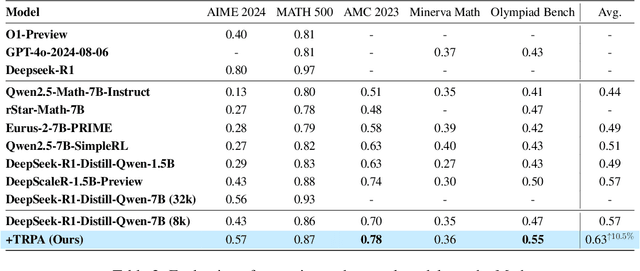
Recently, Large Language Models (LLMs) have rapidly evolved, approaching Artificial General Intelligence (AGI) while benefiting from large-scale reinforcement learning to enhance Human Alignment (HA) and Reasoning. Recent reward-based optimization algorithms, such as Proximal Policy Optimization (PPO) and Group Relative Policy Optimization (GRPO) have achieved significant performance on reasoning tasks, whereas preference-based optimization algorithms such as Direct Preference Optimization (DPO) significantly improve the performance of LLMs on human alignment. However, despite the strong performance of reward-based optimization methods in alignment tasks , they remain vulnerable to reward hacking. Furthermore, preference-based algorithms (such as Online DPO) haven't yet matched the performance of reward-based optimization algorithms (like PPO) on reasoning tasks, making their exploration in this specific area still a worthwhile pursuit. Motivated by these challenges, we propose the Trust Region Preference Approximation (TRPA) algorithm, which integrates rule-based optimization with preference-based optimization for reasoning tasks. As a preference-based algorithm, TRPA naturally eliminates the reward hacking issue. TRPA constructs preference levels using predefined rules, forms corresponding preference pairs, and leverages a novel optimization algorithm for RL training with a theoretical monotonic improvement guarantee. Experimental results demonstrate that TRPA not only achieves competitive performance on reasoning tasks but also exhibits robust stability. The code of this paper are released and updating on https://github.com/XueruiSu/Trust-Region-Preference-Approximation.git.
TeleAntiFraud-28k: An Audio-Text Slow-Thinking Dataset for Telecom Fraud Detection
Apr 01, 2025



The detection of telecom fraud faces significant challenges due to the lack of high-quality multimodal training data that integrates audio signals with reasoning-oriented textual analysis. To address this gap, we present TeleAntiFraud-28k, the first open-source audio-text slow-thinking dataset specifically designed for automated telecom fraud analysis. Our dataset is constructed through three strategies: (1) Privacy-preserved text-truth sample generation using automatically speech recognition (ASR)-transcribed call recordings (with anonymized original audio), ensuring real-world consistency through text-to-speech (TTS) model regeneration; (2) Semantic enhancement via large language model (LLM)-based self-instruction sampling on authentic ASR outputs to expand scenario coverage; (3) Multi-agent adversarial synthesis that simulates emerging fraud tactics through predefined communication scenarios and fraud typologies. The generated dataset contains 28,511 rigorously processed speech-text pairs, complete with detailed annotations for fraud reasoning. The dataset is divided into three tasks: scenario classification, fraud detection, fraud type classification. Furthermore, we construct TeleAntiFraud-Bench, a standardized evaluation benchmark comprising proportionally sampled instances from the dataset, to facilitate systematic testing of model performance on telecom fraud detection tasks. We also contribute a production-optimized supervised fine-tuning (SFT) model trained on hybrid real/synthetic data, while open-sourcing the data processing framework to enable community-driven dataset expansion. This work establishes a foundational framework for multimodal anti-fraud research while addressing critical challenges in data privacy and scenario diversity. The project will be released at https://github.com/JimmyMa99/TeleAntiFraud.
SARChat-Bench-2M: A Multi-Task Vision-Language Benchmark for SAR Image Interpretation
Feb 12, 2025



In the field of synthetic aperture radar (SAR) remote sensing image interpretation, although Vision language models (VLMs) have made remarkable progress in natural language processing and image understanding, their applications remain limited in professional domains due to insufficient domain expertise. This paper innovatively proposes the first large-scale multimodal dialogue dataset for SAR images, named SARChat-2M, which contains approximately 2 million high-quality image-text pairs, encompasses diverse scenarios with detailed target annotations. This dataset not only supports several key tasks such as visual understanding and object detection tasks, but also has unique innovative aspects: this study develop a visual-language dataset and benchmark for the SAR domain, enabling and evaluating VLMs' capabilities in SAR image interpretation, which provides a paradigmatic framework for constructing multimodal datasets across various remote sensing vertical domains. Through experiments on 16 mainstream VLMs, the effectiveness of the dataset has been fully verified, and the first multi-task dialogue benchmark in the SAR field has been successfully established. The project will be released at https://github.com/JimmyMa99/SARChat, aiming to promote the in-depth development and wide application of SAR visual language models.
Reveal the Mystery of DPO: The Connection between DPO and RL Algorithms
Feb 05, 2025With the rapid development of Large Language Models (LLMs), numerous Reinforcement Learning from Human Feedback (RLHF) algorithms have been introduced to improve model safety and alignment with human preferences. These algorithms can be divided into two main frameworks based on whether they require an explicit reward (or value) function for training: actor-critic-based Proximal Policy Optimization (PPO) and alignment-based Direct Preference Optimization (DPO). The mismatch between DPO and PPO, such as DPO's use of a classification loss driven by human-preferred data, has raised confusion about whether DPO should be classified as a Reinforcement Learning (RL) algorithm. To address these ambiguities, we focus on three key aspects related to DPO, RL, and other RLHF algorithms: (1) the construction of the loss function; (2) the target distribution at which the algorithm converges; (3) the impact of key components within the loss function. Specifically, we first establish a unified framework named UDRRA connecting these algorithms based on the construction of their loss functions. Next, we uncover their target policy distributions within this framework. Finally, we investigate the critical components of DPO to understand their impact on the convergence rate. Our work provides a deeper understanding of the relationship between DPO, RL, and other RLHF algorithms, offering new insights for improving existing algorithms.
Language Models as Continuous Self-Evolving Data Engineers
Dec 19, 2024



Large Language Models (LLMs) have demonstrated remarkable capabilities on various tasks, while the further evolvement is limited to the lack of high-quality training data. In addition, traditional training approaches rely too much on expert-labeled data, setting an upper limit on the performance of LLMs. To address this issue, we propose a novel paradigm that enables LLMs to train itself by autonomously generating, cleaning, reviewing, and annotating data with preference information, named LANCE. Our approach demonstrates that LLMs can serve as continuous self-evolving data engineers, significantly reducing the time and cost of the post-training data construction process. Through iterative fine-tuning on different variants of the Qwen2, we validate the effectiveness of LANCE across various tasks, showing that it can continuously improve model performance and maintain high-quality data generation. Across eight benchmark dimensions, LANCE resulted in an average score enhancement of 3.36 for Qwen2-7B and 2.70 for Qwen2-7B-Instruct. This training paradigm with autonomous data construction not only reduces the reliance on human experts or external models but also ensures that the data aligns with human values and preferences, paving the way for the development of future superintelligent systems that can exceed human capabilities.
Molecule Joint Auto-Encoding: Trajectory Pretraining with 2D and 3D Diffusion
Dec 06, 2023Recently, artificial intelligence for drug discovery has raised increasing interest in both machine learning and chemistry domains. The fundamental building block for drug discovery is molecule geometry and thus, the molecule's geometrical representation is the main bottleneck to better utilize machine learning techniques for drug discovery. In this work, we propose a pretraining method for molecule joint auto-encoding (MoleculeJAE). MoleculeJAE can learn both the 2D bond (topology) and 3D conformation (geometry) information, and a diffusion process model is applied to mimic the augmented trajectories of such two modalities, based on which, MoleculeJAE will learn the inherent chemical structure in a self-supervised manner. Thus, the pretrained geometrical representation in MoleculeJAE is expected to benefit downstream geometry-related tasks. Empirically, MoleculeJAE proves its effectiveness by reaching state-of-the-art performance on 15 out of 20 tasks by comparing it with 12 competitive baselines.
Elastic Information Bottleneck
Nov 07, 2023Information bottleneck is an information-theoretic principle of representation learning that aims to learn a maximally compressed representation that preserves as much information about labels as possible. Under this principle, two different methods have been proposed, i.e., information bottleneck (IB) and deterministic information bottleneck (DIB), and have gained significant progress in explaining the representation mechanisms of deep learning algorithms. However, these theoretical and empirical successes are only valid with the assumption that training and test data are drawn from the same distribution, which is clearly not satisfied in many real-world applications. In this paper, we study their generalization abilities within a transfer learning scenario, where the target error could be decomposed into three components, i.e., source empirical error, source generalization gap (SG), and representation discrepancy (RD). Comparing IB and DIB on these terms, we prove that DIB's SG bound is tighter than IB's while DIB's RD is larger than IB's. Therefore, it is difficult to tell which one is better. To balance the trade-off between SG and the RD, we propose an elastic information bottleneck (EIB) to interpolate between the IB and DIB regularizers, which guarantees a Pareto frontier within the IB framework. Additionally, simulations and real data experiments show that EIB has the ability to achieve better domain adaptation results than IB and DIB, which validates the correctness of our theories.