 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrequency-Forcing: From Scaling-as-Time to Soft Frequency Guidance
Apr 21, 2026While standard flow-matching models transport noise to data uniformly, incorporating an explicit generation order - specifically, establishing coarse, low-frequency structure before fine detail - has proven highly effective for synthesizing natural images. Two recent works offer distinct paradigms for this. K-Flow imposes a hard frequency constraint by reinterpreting a frequency scaling variable as flow time, running the trajectory inside a transformed amplitude space. Latent Forcing provides a soft ordering mechanism by coupling the pixel flow with an auxiliary semantic latent flow via asynchronous time schedules, leaving the pixel interpolation path itself untouched. Viewed from the angle of improving pixel generation, we observe that forcing - guiding generation with an earlier-maturing auxiliary stream - offers a highly compatible route to scale-ordered generation without rewriting the core flow coordinate. Building on this, we propose Frequency-Forcing, which realizes K-Flow's frequency ordering through Latent Forcing's soft mechanism: a standard pixel flow is guided by an auxiliary low-frequency stream that matures earlier in time. Unlike Latent Forcing, whose scratchpad relies on a heavy pretrained encoder (e.g., DINO), our frequency scratchpad is derived from the data itself via a lightweight learnable wavelet packet transform. We term this a self-forcing signal, which avoids external dependencies while learning a basis better adapted to data statistics than the fixed bases used in hard frequency flows. On ImageNet-256, Frequency-Forcing consistently improves FID over strong pixel- and latent-space baselines, and naturally composes with a semantic stream to yield further gains. This illustrates that forcing-based scale ordering is a versatile, path-preserving alternative to hard frequency flows.
InertialAR: Autoregressive 3D Molecule Generation with Inertial Frames
Oct 31, 2025Transformer-based autoregressive models have emerged as a unifying paradigm across modalities such as text and images, but their extension to 3D molecule generation remains underexplored. The gap stems from two fundamental challenges: (1) tokenizing molecules into a canonical 1D sequence of tokens that is invariant to both SE(3) transformations and atom index permutations, and (2) designing an architecture capable of modeling hybrid atom-based tokens that couple discrete atom types with continuous 3D coordinates. To address these challenges, we introduce InertialAR. InertialAR devises a canonical tokenization that aligns molecules to their inertial frames and reorders atoms to ensure SE(3) and permutation invariance. Moreover, InertialAR equips the attention mechanism with geometric awareness via geometric rotary positional encoding (GeoRoPE). In addition, it utilizes a hierarchical autoregressive paradigm to predict the next atom-based token, predicting the atom type first and then its 3D coordinates via Diffusion loss. Experimentally, InertialAR achieves state-of-the-art performance on 7 of the 10 evaluation metrics for unconditional molecule generation across QM9, GEOM-Drugs, and B3LYP. Moreover, it significantly outperforms strong baselines in controllable generation for targeted chemical functionality, attaining state-of-the-art results across all 5 metrics.
Flow Along the K-Amplitude for Generative Modeling
Apr 27, 2025In this work, we propose a novel generative learning paradigm, K-Flow, an algorithm that flows along the $K$-amplitude. Here, $k$ is a scaling parameter that organizes frequency bands (or projected coefficients), and amplitude describes the norm of such projected coefficients. By incorporating the $K$-amplitude decomposition, K-Flow enables flow matching across the scaling parameter as time. We discuss three venues and six properties of K-Flow, from theoretical foundations, energy and temporal dynamics, and practical applications, respectively. Specifically, from the practical usage perspective, K-Flow allows steerable generation by controlling the information at different scales. To demonstrate the effectiveness of K-Flow, we conduct experiments on unconditional image generation, class-conditional image generation, and molecule assembly generation. Additionally, we conduct three ablation studies to demonstrate how K-Flow steers scaling parameter to effectively control the resolution of image generation.
GDiffRetro: Retrosynthesis Prediction with Dual Graph Enhanced Molecular Representation and Diffusion Generation
Jan 14, 2025



Retrosynthesis prediction focuses on identifying reactants capable of synthesizing a target product. Typically, the retrosynthesis prediction involves two phases: Reaction Center Identification and Reactant Generation. However, we argue that most existing methods suffer from two limitations in the two phases: (i) Existing models do not adequately capture the ``face'' information in molecular graphs for the reaction center identification. (ii) Current approaches for the reactant generation predominantly use sequence generation in a 2D space, which lacks versatility in generating reasonable distributions for completed reactive groups and overlooks molecules' inherent 3D properties. To overcome the above limitations, we propose GDiffRetro. For the reaction center identification, GDiffRetro uniquely integrates the original graph with its corresponding dual graph to represent molecular structures, which helps guide the model to focus more on the faces in the graph. For the reactant generation, GDiffRetro employs a conditional diffusion model in 3D to further transform the obtained synthon into a complete reactant. Our experimental findings reveal that GDiffRetro outperforms state-of-the-art semi-template models across various evaluative metrics.
AlphaNet: Scaling Up Local Frame-based Atomistic Foundation Model
Jan 13, 2025
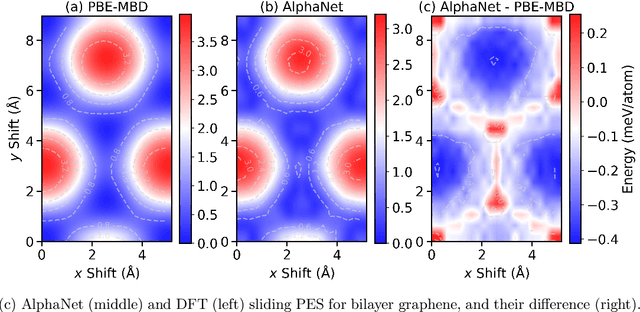
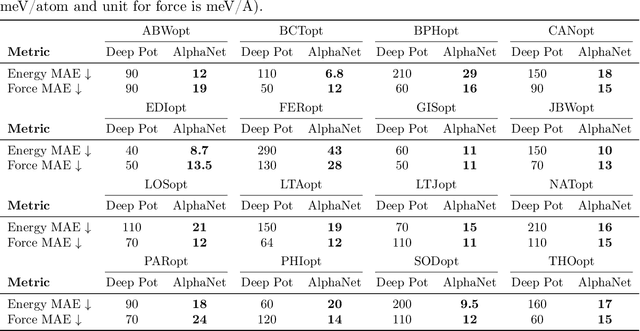
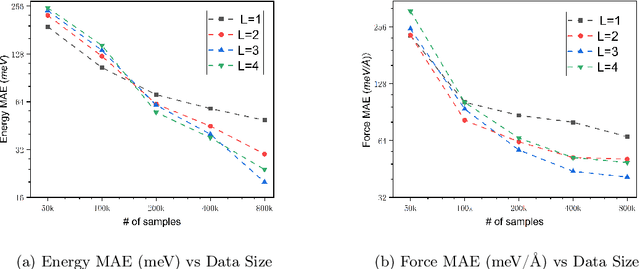
We present AlphaNet, a local frame-based equivariant model designed to achieve both accurate and efficient simulations for atomistic systems. Recently, machine learning force fields (MLFFs) have gained prominence in molecular dynamics simulations due to their advantageous efficiency-accuracy balance compared to classical force fields and quantum mechanical calculations, alongside their transferability across various systems. Despite the advancements in improving model accuracy, the efficiency and scalability of MLFFs remain significant obstacles in practical applications. AlphaNet enhances computational efficiency and accuracy by leveraging the local geometric structures of atomic environments through the construction of equivariant local frames and learnable frame transitions. We substantiate the efficacy of AlphaNet across diverse datasets, including defected graphene, formate decomposition, zeolites, and surface reactions. AlphaNet consistently surpasses well-established models, such as NequIP and DeepPot, in terms of both energy and force prediction accuracy. Notably, AlphaNet offers one of the best trade-offs between computational efficiency and accuracy among existing models. Moreover, AlphaNet exhibits scalability across a broad spectrum of system and dataset sizes, affirming its versatility.
Manifold-Constrained Nucleus-Level Denoising Diffusion Model for Structure-Based Drug Design
Sep 16, 2024Artificial intelligence models have shown great potential in structure-based drug design, generating ligands with high binding affinities. However, existing models have often overlooked a crucial physical constraint: atoms must maintain a minimum pairwise distance to avoid separation violation, a phenomenon governed by the balance of attractive and repulsive forces. To mitigate such separation violations, we propose NucleusDiff. It models the interactions between atomic nuclei and their surrounding electron clouds by enforcing the distance constraint between the nuclei and manifolds. We quantitatively evaluate NucleusDiff using the CrossDocked2020 dataset and a COVID-19 therapeutic target, demonstrating that NucleusDiff reduces violation rate by up to 100.00% and enhances binding affinity by up to 22.16%, surpassing state-of-the-art models for structure-based drug design. We also provide qualitative analysis through manifold sampling, visually confirming the effectiveness of NucleusDiff in reducing separation violations and improving binding affinities.
Sculpting Molecules in 3D: A Flexible Substructure Aware Framework for Text-Oriented Molecular Optimization
Mar 06, 2024



The integration of deep learning, particularly AI-Generated Content, with high-quality data derived from ab initio calculations has emerged as a promising avenue for transforming the landscape of scientific research. However, the challenge of designing molecular drugs or materials that incorporate multi-modality prior knowledge remains a critical and complex undertaking. Specifically, achieving a practical molecular design necessitates not only meeting the diversity requirements but also addressing structural and textural constraints with various symmetries outlined by domain experts. In this article, we present an innovative approach to tackle this inverse design problem by formulating it as a multi-modality guidance generation/optimization task. Our proposed solution involves a textural-structure alignment symmetric diffusion framework for the implementation of molecular generation/optimization tasks, namely 3DToMolo. 3DToMolo aims to harmonize diverse modalities, aligning them seamlessly to produce molecular structures adhere to specified symmetric structural and textural constraints by experts in the field. Experimental trials across three guidance generation settings have shown a superior hit generation performance compared to state-of-the-art methodologies. Moreover, 3DToMolo demonstrates the capability to generate novel molecules, incorporating specified target substructures, without the need for prior knowledge. This work not only holds general significance for the advancement of deep learning methodologies but also paves the way for a transformative shift in molecular design strategies. 3DToMolo creates opportunities for a more nuanced and effective exploration of the vast chemical space, opening new frontiers in the development of molecular entities with tailored properties and functionalities.
A Multi-Grained Symmetric Differential Equation Model for Learning Protein-Ligand Binding Dynamics
Feb 01, 2024In drug discovery, molecular dynamics (MD) simulation for protein-ligand binding provides a powerful tool for predicting binding affinities, estimating transport properties, and exploring pocket sites. There has been a long history of improving the efficiency of MD simulations through better numerical methods and, more recently, by utilizing machine learning (ML) methods. Yet, challenges remain, such as accurate modeling of extended-timescale simulations. To address this issue, we propose NeuralMD, the first ML surrogate that can facilitate numerical MD and provide accurate simulations in protein-ligand binding. We propose a principled approach that incorporates a novel physics-informed multi-grained group symmetric framework. Specifically, we propose (1) a BindingNet model that satisfies group symmetry using vector frames and captures the multi-level protein-ligand interactions, and (2) an augmented neural differential equation solver that learns the trajectory under Newtonian mechanics. For the experiment, we design ten single-trajectory and three multi-trajectory binding simulation tasks. We show the efficiency and effectiveness of NeuralMD, with a 2000$\times$ speedup over standard numerical MD simulation and outperforming all other ML approaches by up to 80% under the stability metric. We further qualitatively show that NeuralMD reaches more stable binding predictions compared to other machine learning methods.
A quatum inspired neural network for geometric modeling
Jan 03, 2024By conceiving physical systems as 3D many-body point clouds, geometric graph neural networks (GNNs), such as SE(3)/E(3) equivalent GNNs, have showcased promising performance. In particular, their effective message-passing mechanics make them adept at modeling molecules and crystalline materials. However, current geometric GNNs only offer a mean-field approximation of the many-body system, encapsulated within two-body message passing, thus falling short in capturing intricate relationships within these geometric graphs. To address this limitation, tensor networks, widely employed by computational physics to handle manybody systems using high-order tensors, have been introduced. Nevertheless, integrating these tensorized networks into the message-passing framework of GNNs faces scalability and symmetry conservation (e.g., permutation and rotation) challenges. In response, we introduce an innovative equivariant Matrix Product State (MPS)-based message-passing strategy, through achieving an efficient implementation of the tensor contraction operation. Our method effectively models complex many-body relationships, suppressing mean-field approximations, and captures symmetries within geometric graphs. Importantly, it seamlessly replaces the standard message-passing and layer-aggregation modules intrinsic to geometric GNNs. We empirically validate the superior accuracy of our approach on benchmark tasks, including predicting classical Newton systems and quantum tensor Hamiltonian matrices. To our knowledge, our approach represents the inaugural utilization of parameterized geometric tensor networks.
Molecule Joint Auto-Encoding: Trajectory Pretraining with 2D and 3D Diffusion
Dec 06, 2023Recently, artificial intelligence for drug discovery has raised increasing interest in both machine learning and chemistry domains. The fundamental building block for drug discovery is molecule geometry and thus, the molecule's geometrical representation is the main bottleneck to better utilize machine learning techniques for drug discovery. In this work, we propose a pretraining method for molecule joint auto-encoding (MoleculeJAE). MoleculeJAE can learn both the 2D bond (topology) and 3D conformation (geometry) information, and a diffusion process model is applied to mimic the augmented trajectories of such two modalities, based on which, MoleculeJAE will learn the inherent chemical structure in a self-supervised manner. Thus, the pretrained geometrical representation in MoleculeJAE is expected to benefit downstream geometry-related tasks. Empirically, MoleculeJAE proves its effectiveness by reaching state-of-the-art performance on 15 out of 20 tasks by comparing it with 12 competitive baselines.