 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBuilding-Block Aware Generative Modeling for 3D Crystals of Metal Organic Frameworks
May 13, 2025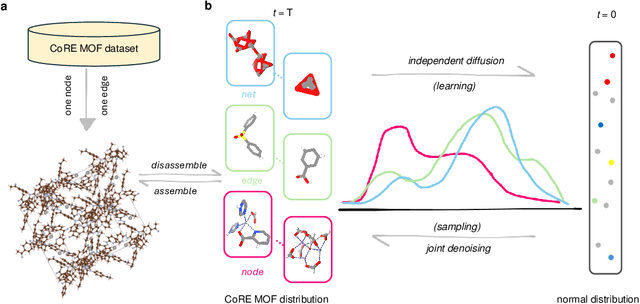
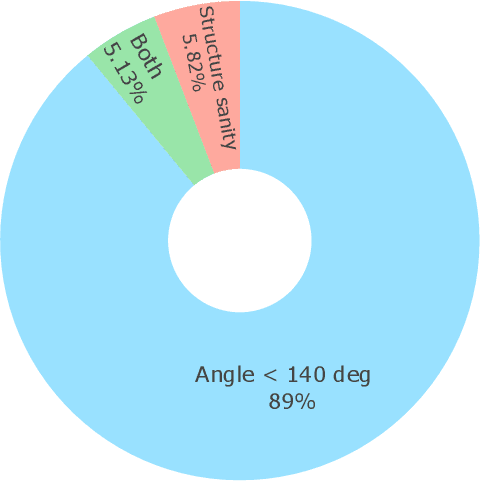
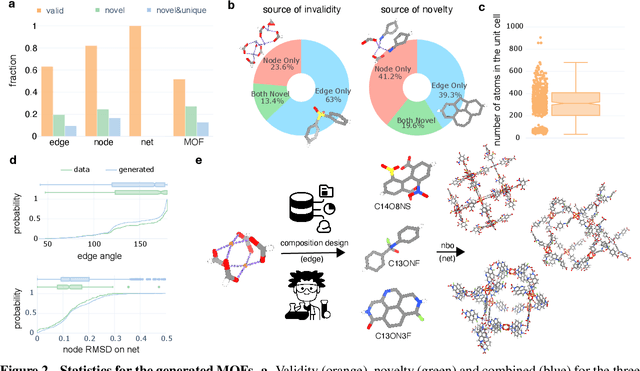
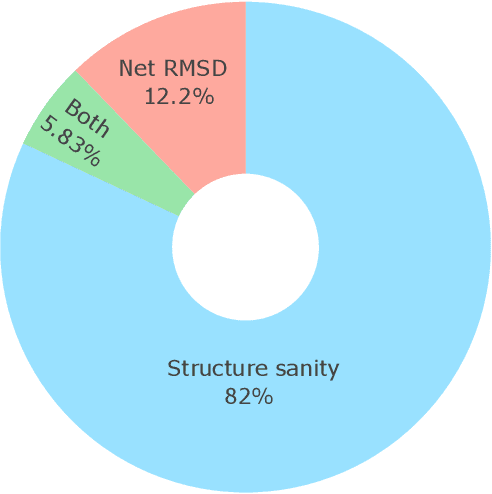
Metal-organic frameworks (MOFs) marry inorganic nodes, organic edges, and topological nets into programmable porous crystals, yet their astronomical design space defies brute-force synthesis. Generative modeling holds ultimate promise, but existing models either recycle known building blocks or are restricted to small unit cells. We introduce Building-Block-Aware MOF Diffusion (BBA MOF Diffusion), an SE(3)-equivariant diffusion model that learns 3D all-atom representations of individual building blocks, encoding crystallographic topological nets explicitly. Trained on the CoRE-MOF database, BBA MOF Diffusion readily samples MOFs with unit cells containing 1000 atoms with great geometric validity, novelty, and diversity mirroring experimental databases. Its native building-block representation produces unprecedented metal nodes and organic edges, expanding accessible chemical space by orders of magnitude. One high-scoring [Zn(1,4-TDC)(EtOH)2] MOF predicted by the model was synthesized, where powder X-ray diffraction, thermogravimetric analysis, and N2 sorption confirm its structural fidelity. BBA-Diff thus furnishes a practical pathway to synthesizable and high-performing MOFs.
Large Language Models Are Innate Crystal Structure Generators
Feb 28, 2025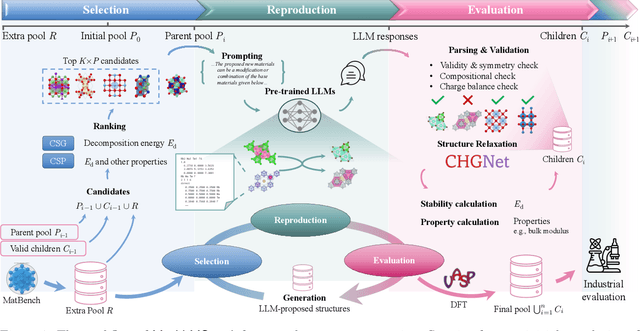
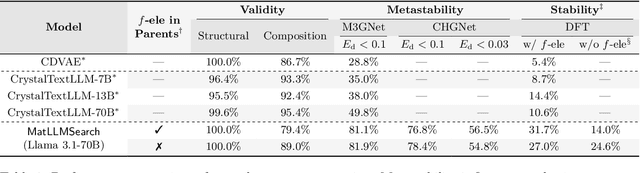
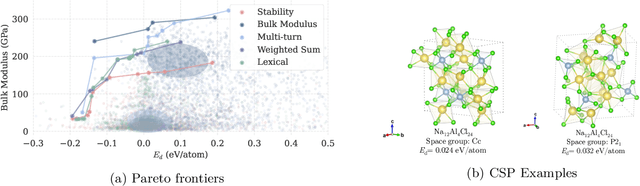
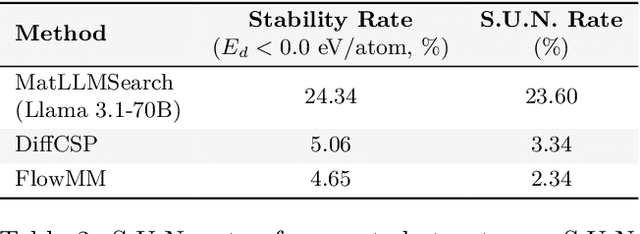
Crystal structure generation is fundamental to materials discovery, enabling the prediction of novel materials with desired properties. While existing approaches leverage Large Language Models (LLMs) through extensive fine-tuning on materials databases, we show that pre-trained LLMs can inherently generate stable crystal structures without additional training. Our novel framework MatLLMSearch integrates pre-trained LLMs with evolutionary search algorithms, achieving a 78.38% metastable rate validated by machine learning interatomic potentials and 31.7% DFT-verified stability via quantum mechanical calculations, outperforming specialized models such as CrystalTextLLM. Beyond crystal structure generation, we further demonstrate that our framework can be readily adapted to diverse materials design tasks, including crystal structure prediction and multi-objective optimization of properties such as deformation energy and bulk modulus, all without fine-tuning. These results establish pre-trained LLMs as versatile and effective tools for materials discovery, opening up new venues for crystal structure generation with reduced computational overhead and broader accessibility.
AlphaNet: Scaling Up Local Frame-based Atomistic Foundation Model
Jan 13, 2025
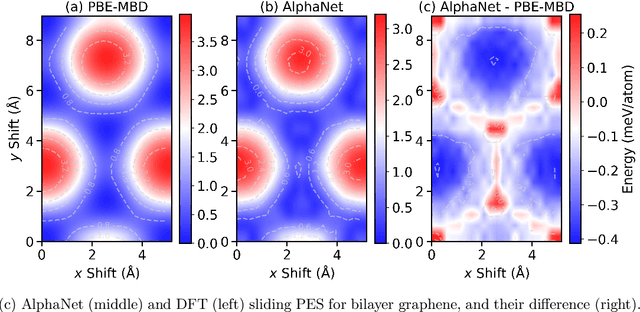
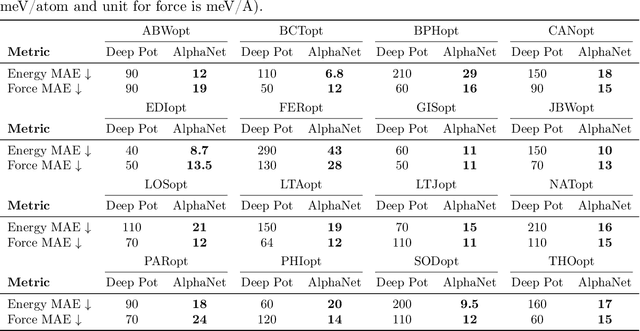
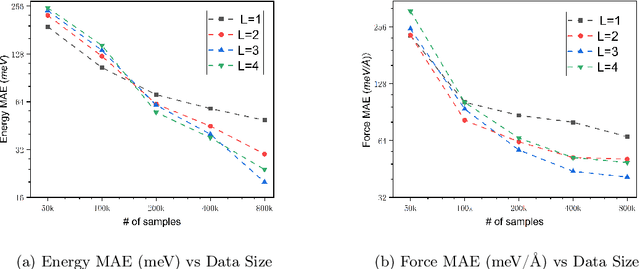
We present AlphaNet, a local frame-based equivariant model designed to achieve both accurate and efficient simulations for atomistic systems. Recently, machine learning force fields (MLFFs) have gained prominence in molecular dynamics simulations due to their advantageous efficiency-accuracy balance compared to classical force fields and quantum mechanical calculations, alongside their transferability across various systems. Despite the advancements in improving model accuracy, the efficiency and scalability of MLFFs remain significant obstacles in practical applications. AlphaNet enhances computational efficiency and accuracy by leveraging the local geometric structures of atomic environments through the construction of equivariant local frames and learnable frame transitions. We substantiate the efficacy of AlphaNet across diverse datasets, including defected graphene, formate decomposition, zeolites, and surface reactions. AlphaNet consistently surpasses well-established models, such as NequIP and DeepPot, in terms of both energy and force prediction accuracy. Notably, AlphaNet offers one of the best trade-offs between computational efficiency and accuracy among existing models. Moreover, AlphaNet exhibits scalability across a broad spectrum of system and dataset sizes, affirming its versatility.
Generative Design of Functional Metal Complexes Utilizing the Internal Knowledge of Large Language Models
Oct 21, 2024


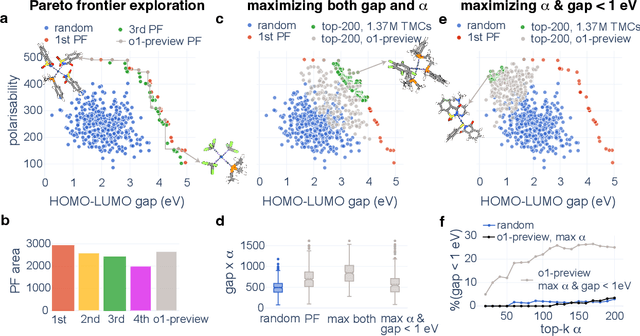
Designing functional transition metal complexes (TMCs) faces challenges due to the vast search space of metals and ligands, requiring efficient optimization strategies. Traditional genetic algorithms (GAs) are commonly used, employing random mutations and crossovers driven by explicit mathematical objectives to explore this space. Transferring knowledge between different GA tasks, however, is difficult. We integrate large language models (LLMs) into the evolutionary optimization framework (LLM-EO) and apply it in both single- and multi-objective optimization for TMCs. We find that LLM-EO surpasses traditional GAs by leveraging the chemical knowledge of LLMs gained during their extensive pretraining. Remarkably, without supervised fine-tuning, LLMs utilize the full historical data from optimization processes, outperforming those focusing only on top-performing TMCs. LLM-EO successfully identifies eight of the top-20 TMCs with the largest HOMO-LUMO gaps by proposing only 200 candidates out of a 1.37 million TMCs space. Through prompt engineering using natural language, LLM-EO introduces unparalleled flexibility into multi-objective optimizations, thereby circumventing the necessity for intricate mathematical formulations. As generative models, LLMs can suggest new ligands and TMCs with unique properties by merging both internal knowledge and external chemistry data, thus combining the benefits of efficient optimization and molecular generation. With increasing potential of LLMs as pretrained foundational models and new post-training inference strategies, we foresee broad applications of LLM-based evolutionary optimization in chemistry and materials design.
Doob's Lagrangian: A Sample-Efficient Variational Approach to Transition Path Sampling
Oct 10, 2024

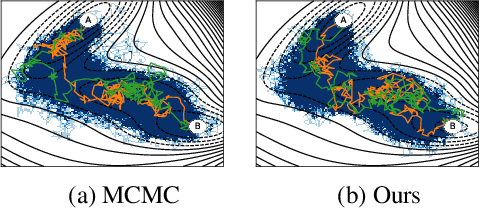
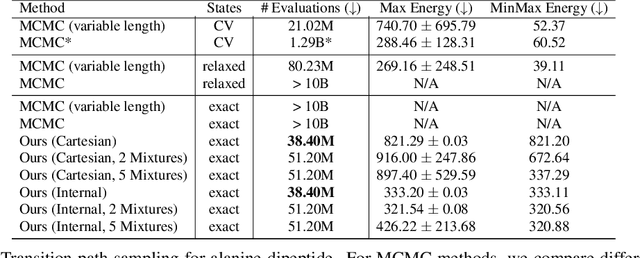
Rare event sampling in dynamical systems is a fundamental problem arising in the natural sciences, which poses significant computational challenges due to an exponentially large space of trajectories. For settings where the dynamical system of interest follows a Brownian motion with known drift, the question of conditioning the process to reach a given endpoint or desired rare event is definitively answered by Doob's h-transform. However, the naive estimation of this transform is infeasible, as it requires simulating sufficiently many forward trajectories to estimate rare event probabilities. In this work, we propose a variational formulation of Doob's $h$-transform as an optimization problem over trajectories between a given initial point and the desired ending point. To solve this optimization, we propose a simulation-free training objective with a model parameterization that imposes the desired boundary conditions by design. Our approach significantly reduces the search space over trajectories and avoids expensive trajectory simulation and inefficient importance sampling estimators which are required in existing methods. We demonstrate the ability of our method to find feasible transition paths on real-world molecular simulation and protein folding tasks.
Efficient Evolutionary Search Over Chemical Space with Large Language Models
Jun 23, 2024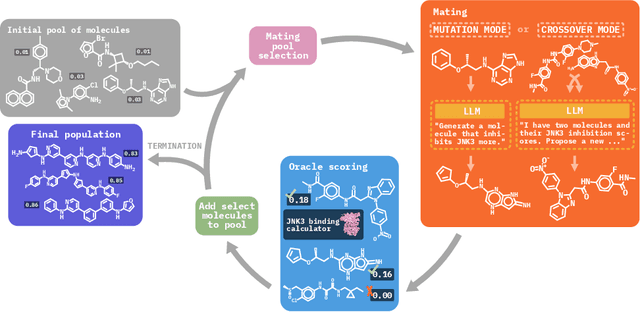
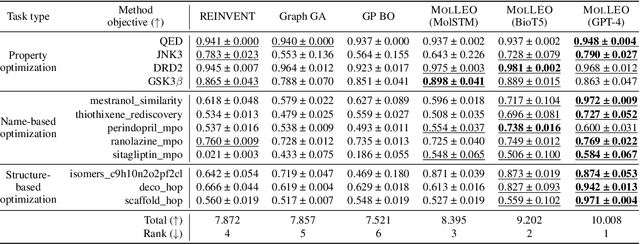
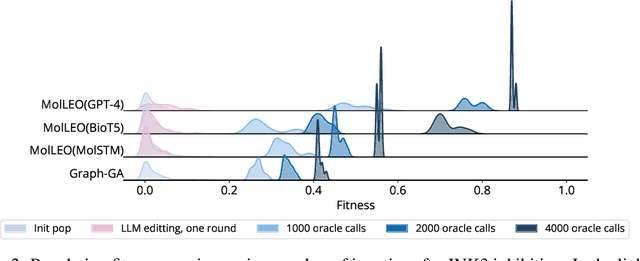
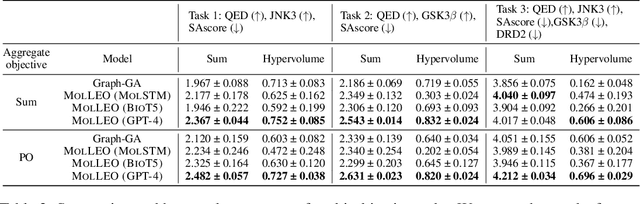
Molecular discovery, when formulated as an optimization problem, presents significant computational challenges because optimization objectives can be non-differentiable. Evolutionary Algorithms (EAs), often used to optimize black-box objectives in molecular discovery, traverse chemical space by performing random mutations and crossovers, leading to a large number of expensive objective evaluations. In this work, we ameliorate this shortcoming by incorporating chemistry-aware Large Language Models (LLMs) into EAs. Namely, we redesign crossover and mutation operations in EAs using LLMs trained on large corpora of chemical information. We perform extensive empirical studies on both commercial and open-source models on multiple tasks involving property optimization, molecular rediscovery, and structure-based drug design, demonstrating that the joint usage of LLMs with EAs yields superior performance over all baseline models across single- and multi-objective settings. We demonstrate that our algorithm improves both the quality of the final solution and convergence speed, thereby reducing the number of required objective evaluations. Our code is available at http://github.com/zoom-wang112358/MOLLEO
Navigating Chemical Space with Latent Flows
May 08, 2024



Recent progress of deep generative models in the vision and language domain has stimulated significant interest in more structured data generation such as molecules. However, beyond generating new random molecules, efficient exploration and a comprehensive understanding of the vast chemical space are of great importance to molecular science and applications in drug design and materials discovery. In this paper, we propose a new framework, ChemFlow, to traverse chemical space through navigating the latent space learned by molecule generative models through flows. We introduce a dynamical system perspective that formulates the problem as learning a vector field that transports the mass of the molecular distribution to the region with desired molecular properties or structure diversity. Under this framework, we unify previous approaches on molecule latent space traversal and optimization and propose alternative competing methods incorporating different physical priors. We validate the efficacy of ChemFlow on molecule manipulation and single- and multi-objective molecule optimization tasks under both supervised and unsupervised molecular discovery settings. Codes and demos are publicly available on GitHub at https://github.com/garywei944/ChemFlow.
React-OT: Optimal Transport for Generating Transition State in Chemical Reactions
Apr 20, 2024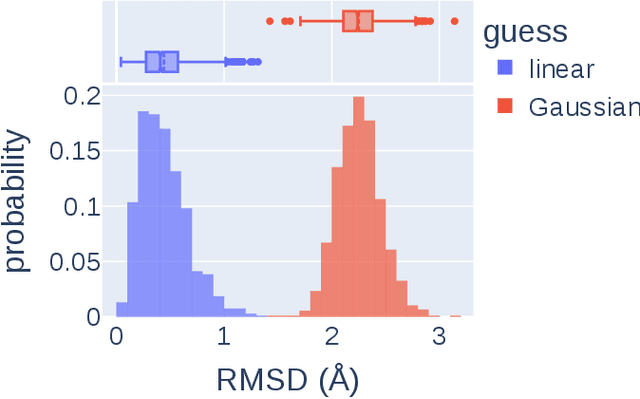
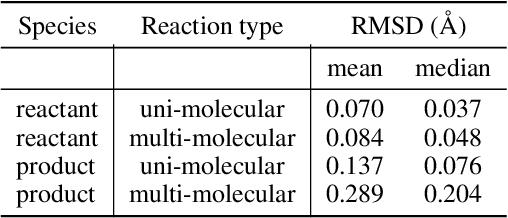
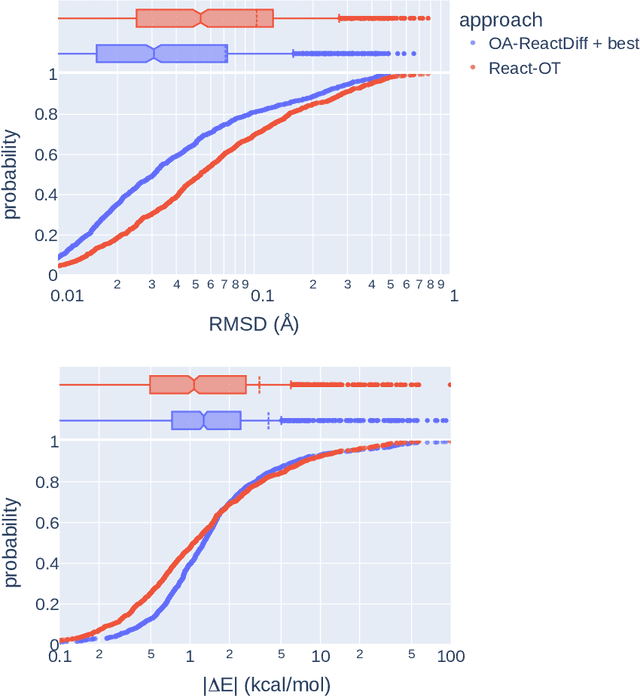
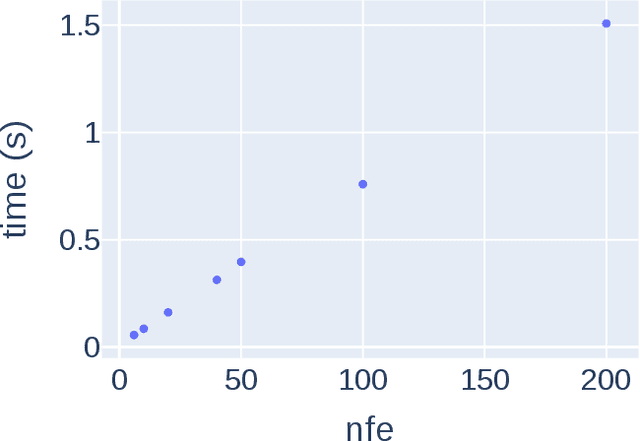
Transition states (TSs) are transient structures that are key in understanding reaction mechanisms and designing catalysts but challenging to be captured in experiments. Alternatively, many optimization algorithms have been developed to search for TSs computationally. Yet the cost of these algorithms driven by quantum chemistry methods (usually density functional theory) is still high, posing challenges for their applications in building large reaction networks for reaction exploration. Here we developed React-OT, an optimal transport approach for generating unique TS structures from reactants and products. React-OT generates highly accurate TS structures with a median structural root mean square deviation (RMSD) of 0.053{\AA} and median barrier height error of 1.06 kcal/mol requiring only 0.4 second per reaction. The RMSD and barrier height error is further improved by roughly 25% through pretraining React-OT on a large reaction dataset obtained with a lower level of theory, GFN2-xTB. We envision the great accuracy and fast inference of React-OT useful in targeting TSs when exploring chemical reactions with unknown mechanisms.
Symmetry-Informed Geometric Representation for Molecules, Proteins, and Crystalline Materials
Jun 15, 2023



Artificial intelligence for scientific discovery has recently generated significant interest within the machine learning and scientific communities, particularly in the domains of chemistry, biology, and material discovery. For these scientific problems, molecules serve as the fundamental building blocks, and machine learning has emerged as a highly effective and powerful tool for modeling their geometric structures. Nevertheless, due to the rapidly evolving process of the field and the knowledge gap between science (e.g., physics, chemistry, & biology) and machine learning communities, a benchmarking study on geometrical representation for such data has not been conducted. To address such an issue, in this paper, we first provide a unified view of the current symmetry-informed geometric methods, classifying them into three main categories: invariance, equivariance with spherical frame basis, and equivariance with vector frame basis. Then we propose a platform, coined Geom3D, which enables benchmarking the effectiveness of geometric strategies. Geom3D contains 16 advanced symmetry-informed geometric representation models and 14 geometric pretraining methods over 46 diverse datasets, including small molecules, proteins, and crystalline materials. We hope that Geom3D can, on the one hand, eliminate barriers for machine learning researchers interested in exploring scientific problems; and, on the other hand, provide valuable guidance for researchers in computational chemistry, structural biology, and materials science, aiding in the informed selection of representation techniques for specific applications.
Accurate transition state generation with an object-aware equivariant elementary reaction diffusion model
Apr 17, 2023
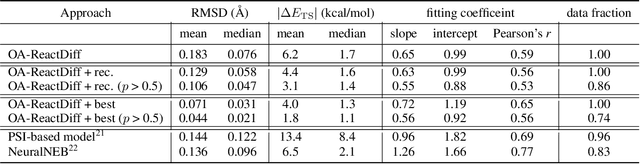
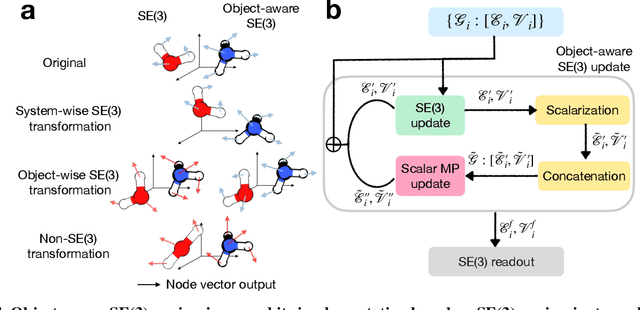
Transition state (TS) search is key in chemistry for elucidating reaction mechanisms and exploring reaction networks. The search for accurate 3D TS structures, however, requires numerous computationally intensive quantum chemistry calculations due to the complexity of potential energy surfaces. Here, we developed an object-aware SE(3) equivariant diffusion model that satisfies all physical symmetries and constraints for generating sets of structures - reactant, TS, and product - in an elementary reaction. Provided reactant and product, this model generates a TS structure in seconds instead of hours required when performing quantum chemistry-based optimizations. The generated TS structures achieve a median of 0.08 {\AA} root mean square deviation compared to the true TS. With a confidence scoring model for uncertainty quantification, we approach an accuracy required for reaction rate estimation (2.6 kcal/mol) by only performing quantum chemistry-based optimizations on 14\% of the most challenging reactions. We envision the proposed approach useful in constructing large reaction networks with unknown mechanisms.