 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInverse design of bespoke interatomic potentials via active learning by information-matching
Jun 06, 2026Interatomic potentials (IPs) enable large-scale atomistic simulations beyond the reach of first-principles methods, but their predictive reliability depends critically on the selection of training data, quantified uncertainty, and model expressiveness. Active learning (AL) provides a principled framework for constructing efficient and accurate IPs, yet most strategies reduce parameter uncertainty without explicitly accounting for the specific material properties being predicted. The information-matching (IM) approach addresses this limitation by requiring that the selected training data provide at least as much parameter space information as needed to achieve prescribed uncertainty targets for selected quantities of interest (QoIs). Here, we apply IM to develop bespoke IPs specifically tailored for predicting plastic strength in metals. Due to the high computational cost of simulating plastic strength, we employ an indirect IM strategy that targets inexpensive intermediate QoIs that correlate with strength. The IM method enables precise parameter constraints with minimal training data, yielding precise predictions for both the intermediate QoIs and plastic strength. Yet, model error remains a key limitation, and a post hoc uncertainty inflation correction provides a viable means to mitigate this limitation. These findings illustrate both the promise and limits of uncertainty-aware AL for predicting complex material properties.
Generative Inversion of Spectroscopic Data for Amorphous Structure Elucidation
Mar 24, 2026Determining atomistic structures from characterization data is one of the most common yet intricate problems in materials science. Particularly in amorphous materials, proposing structures that balance realism and agreement with experiments requires expert guidance, good interatomic potentials, or both. Here, we introduce GLASS, a generative framework that inverts multi-modal spectroscopic measurements into realistic atomistic structures without knowledge of the potential energy surface. A score-based model learns a structural prior from low-fidelity data and samples out-of-distribution structures conditioned on differentiable spectral targets. Reconstructions using pair distribution functions (PDFs), X-ray absorption spectroscopy, and diffraction measurements quantify the complementarity between spectral modalities and demonstrate that PDFs is the most informative probe for our framework. We use GLASS to rationalize three contested experimental problems: paracrystallinity in amorphous silicon, a liquid-liquid phase transition in sulfur, and ball-milled amorphous ice. In each case, generated structures reproduce experimental measurements and reveal mechanisms inaccessible to diffraction analysis alone.
Maximizing Efficiency of Dataset Compression for Machine Learning Potentials With Information Theory
Nov 13, 2025Machine learning interatomic potentials (MLIPs) balance high accuracy and lower costs compared to density functional theory calculations, but their performance often depends on the size and diversity of training datasets. Large datasets improve model accuracy and generalization but are computationally expensive to produce and train on, while smaller datasets risk discarding rare but important atomic environments and compromising MLIP accuracy/reliability. Here, we develop an information-theoretical framework to quantify the efficiency of dataset compression methods and propose an algorithm that maximizes this efficiency. By framing atomistic dataset compression as an instance of the minimum set cover (MSC) problem over atom-centered environments, our method identifies the smallest subset of structures that contains as much information as possible from the original dataset while pruning redundant information. The approach is extensively demonstrated on the GAP-20 and TM23 datasets, and validated on 64 varied datasets from the ColabFit repository. Across all cases, MSC consistently retains outliers, preserves dataset diversity, and reproduces the long-tail distributions of forces even at high compression rates, outperforming other subsampling methods. Furthermore, MLIPs trained on MSC-compressed datasets exhibit reduced error for out-of-distribution data even in low-data regimes. We explain these results using an outlier analysis and show that such quantitative conclusions could not be achieved with conventional dimensionality reduction methods. The algorithm is implemented in the open-source QUESTS package and can be used for several tasks in atomistic modeling, from data subsampling, outlier detection, and training improved MLIPs at a lower cost.
Large Language Models Are Innate Crystal Structure Generators
Feb 28, 2025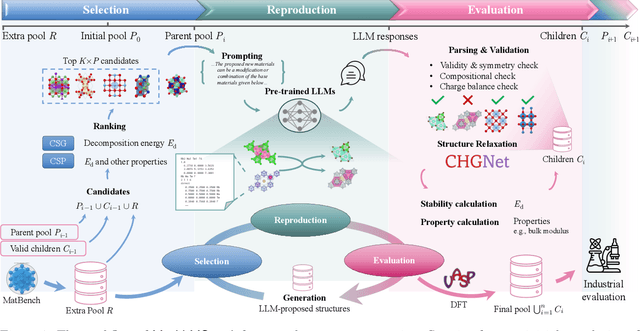
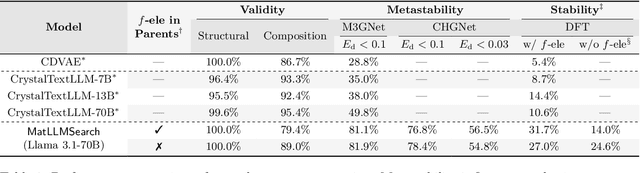
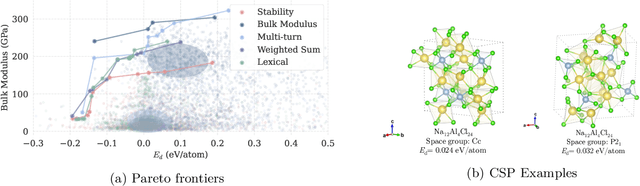
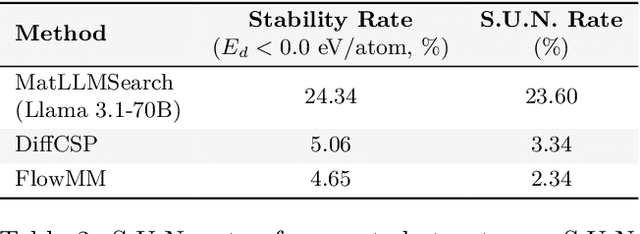
Crystal structure generation is fundamental to materials discovery, enabling the prediction of novel materials with desired properties. While existing approaches leverage Large Language Models (LLMs) through extensive fine-tuning on materials databases, we show that pre-trained LLMs can inherently generate stable crystal structures without additional training. Our novel framework MatLLMSearch integrates pre-trained LLMs with evolutionary search algorithms, achieving a 78.38% metastable rate validated by machine learning interatomic potentials and 31.7% DFT-verified stability via quantum mechanical calculations, outperforming specialized models such as CrystalTextLLM. Beyond crystal structure generation, we further demonstrate that our framework can be readily adapted to diverse materials design tasks, including crystal structure prediction and multi-objective optimization of properties such as deformation energy and bulk modulus, all without fine-tuning. These results establish pre-trained LLMs as versatile and effective tools for materials discovery, opening up new venues for crystal structure generation with reduced computational overhead and broader accessibility.
Information theory unifies atomistic machine learning, uncertainty quantification, and materials thermodynamics
Apr 18, 2024An accurate description of information is relevant for a range of problems in atomistic modeling, such as sampling methods, detecting rare events, analyzing datasets, or performing uncertainty quantification (UQ) in machine learning (ML)-driven simulations. Although individual methods have been proposed for each of these tasks, they lack a common theoretical background integrating their solutions. Here, we introduce an information theoretical framework that unifies predictions of phase transformations, kinetic events, dataset optimality, and model-free UQ from atomistic simulations, thus bridging materials modeling, ML, and statistical mechanics. We first demonstrate that, for a proposed representation, the information entropy of a distribution of atom-centered environments is a surrogate value for thermodynamic entropy. Using molecular dynamics (MD) simulations, we show that information entropy differences from trajectories can be used to build phase diagrams, identify rare events, and recover classical theories of nucleation. Building on these results, we use this general concept of entropy to quantify information in datasets for ML interatomic potentials (IPs), informing compression, explaining trends in testing errors, and evaluating the efficiency of active learning strategies. Finally, we propose a model-free UQ method for MLIPs using information entropy, showing it reliably detects extrapolation regimes, scales to millions of atoms, and goes beyond model errors. This method is made available as the package QUESTS: Quick Uncertainty and Entropy via STructural Similarity, providing a new unifying theory for data-driven atomistic modeling and combining efforts in ML, first-principles thermodynamics, and simulations.
Inorganic synthesis-structure maps in zeolites with machine learning and crystallographic distances
Jul 20, 2023



Zeolites are inorganic materials known for their diversity of applications, synthesis conditions, and resulting polymorphs. Although their synthesis is controlled both by inorganic and organic synthesis conditions, computational studies of zeolite synthesis have focused mostly on organic template design. In this work, we use a strong distance metric between crystal structures and machine learning (ML) to create inorganic synthesis maps in zeolites. Starting with 253 known zeolites, we show how the continuous distances between frameworks reproduce inorganic synthesis conditions from the literature without using labels such as building units. An unsupervised learning analysis shows that neighboring zeolites according to our metric often share similar inorganic synthesis conditions, even in template-based routes. In combination with ML classifiers, we find synthesis-structure relationships for 14 common inorganic conditions in zeolites, namely Al, B, Be, Ca, Co, F, Ga, Ge, K, Mg, Na, P, Si, and Zn. By explaining the model predictions, we demonstrate how (dis)similarities towards known structures can be used as features for the synthesis space. Finally, we show how these methods can be used to predict inorganic synthesis conditions for unrealized frameworks in hypothetical databases and interpret the outcomes by extracting local structural patterns from zeolites. In combination with template design, this work can accelerate the exploration of the space of synthesis conditions for zeolites.
Data efficiency and extrapolation trends in neural network interatomic potentials
Feb 12, 2023Over the last few years, key architectural advances have been proposed for neural network interatomic potentials (NNIPs), such as incorporating message-passing networks, equivariance, or many-body expansion terms. Although modern NNIP models exhibit nearly negligible differences in energy/forces errors, improvements in accuracy are still considered the main target when developing new NNIP architectures. In this work, we investigate how architectural choices influence the trainability and generalization error in NNIPs, revealing trends in extrapolation, data efficiency, and loss landscapes. First, we show that modern NNIP architectures recover the underlying potential energy surface (PES) of the training data even when trained to corrupted labels. Second, generalization metrics such as errors on high-temperature samples from the 3BPA dataset are demonstrated to follow a scaling relation for a variety of models. Thus, improvements in accuracy metrics may not bring independent information on the robust generalization of NNIPs. To circumvent this problem, we relate loss landscapes to model generalization across datasets. Using this probe, we explain why NNIPs with similar accuracy metrics exhibit different abilities to extrapolate and how training to forces improves the optimization landscape of a model. As an example, we show that MACE can predict PESes with reasonable error after being trained to as few as five data points, making it an example of a "few-shot" model for learning PESes. On the other hand, models with similar accuracy metrics such as NequIP show smaller ability to extrapolate in this extremely low-data regime. Our work provides a deep learning justification for the performance of many common NNIPs, and introduces tools beyond accuracy metrics that can be used to inform the development of next-generation models.
Adversarial Attacks on Uncertainty Enable Active Learning for Neural Network Potentials
Feb 01, 2021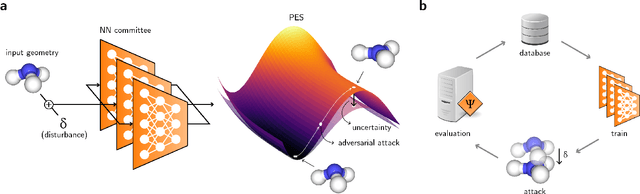
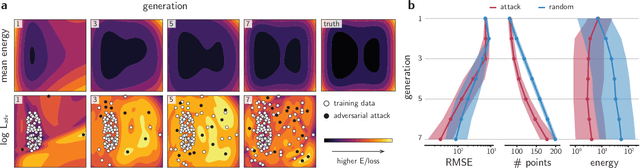
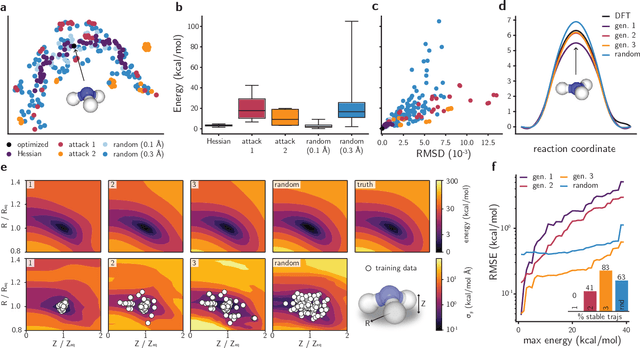
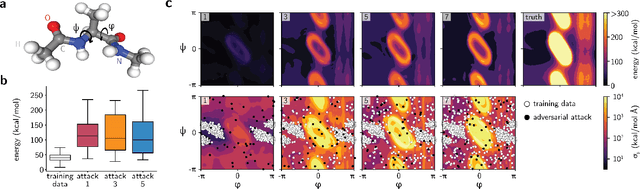
Neural network (NN)-based interatomic potentials provide fast prediction of potential energy surfaces with the accuracy of electronic structure methods. However, NN predictions are only reliable within well-learned training domains, with unknown behavior when extrapolating. Uncertainty quantification through NN committees identify domains with low prediction confidence, but thoroughly exploring the configuration space for training NN potentials often requires slow atomistic simulations. Here, we employ adversarial attacks with a differentiable uncertainty metric to sample new molecular geometries and bootstrap NN potentials. In combination with an active learning loop, the extrapolation power of NN potentials is improved beyond the original training data with few additional samples. The framework is demonstrated on multiple examples, leading to better sampling of kinetic barriers and collective variables without extensive prior data on the relevant geometries. Adversarial attacks are new ways to simultaneously sample the phase space and bootstrap NN potentials, increasing their robustness and enabling a faster, accurate prediction of potential energy landscapes.
Generative Models for Automatic Chemical Design
Jul 02, 2019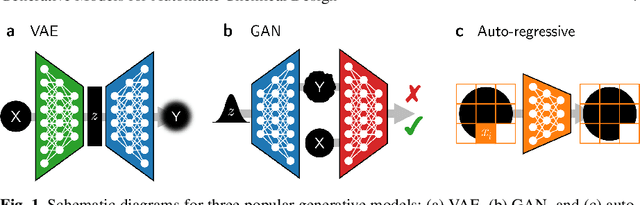
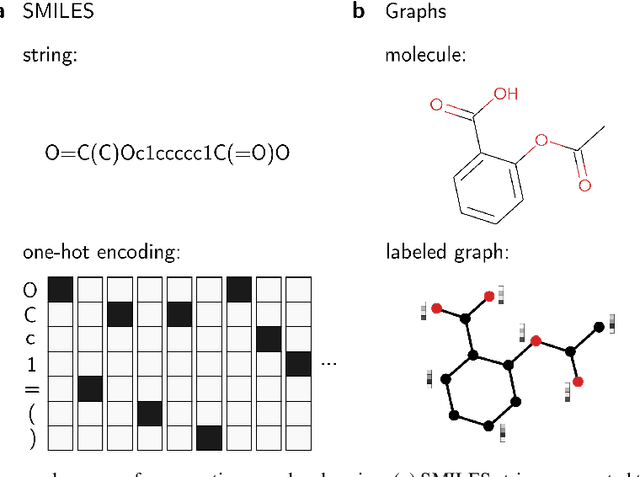
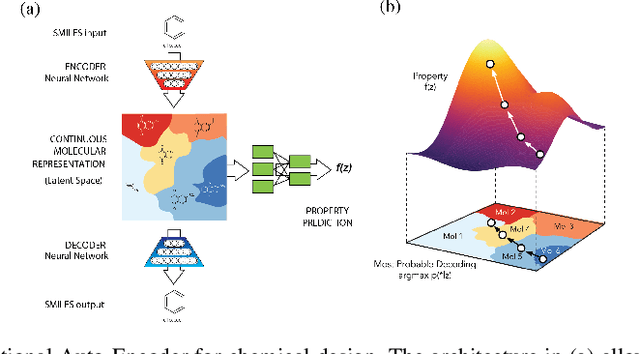
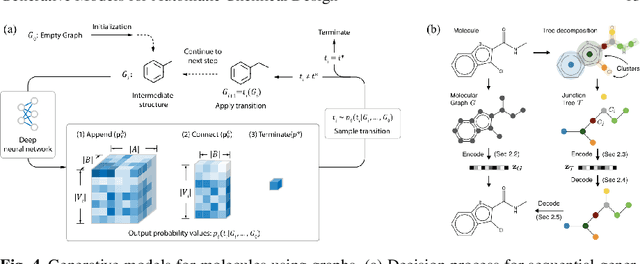
Materials discovery is decisive for tackling urgent challenges related to energy, the environment, health care and many others. In chemistry, conventional methodologies for innovation usually rely on expensive and incremental strategies to optimize properties from molecular structures. On the other hand, inverse approaches map properties to structures, thus expediting the design of novel useful compounds. In this chapter, we examine the way in which current deep generative models are addressing the inverse chemical discovery paradigm. We begin by revisiting early inverse design algorithms. Then, we introduce generative models for molecular systems and categorize them according to their architecture and molecular representation. Using this classification, we review the evolution and performance of important molecular generation schemes reported in the literature. Finally, we conclude highlighting the prospects and challenges of generative models as cutting edge tools in materials discovery.