 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhanced sampling of robust molecular datasets with uncertainty-based collective variables
Feb 06, 2024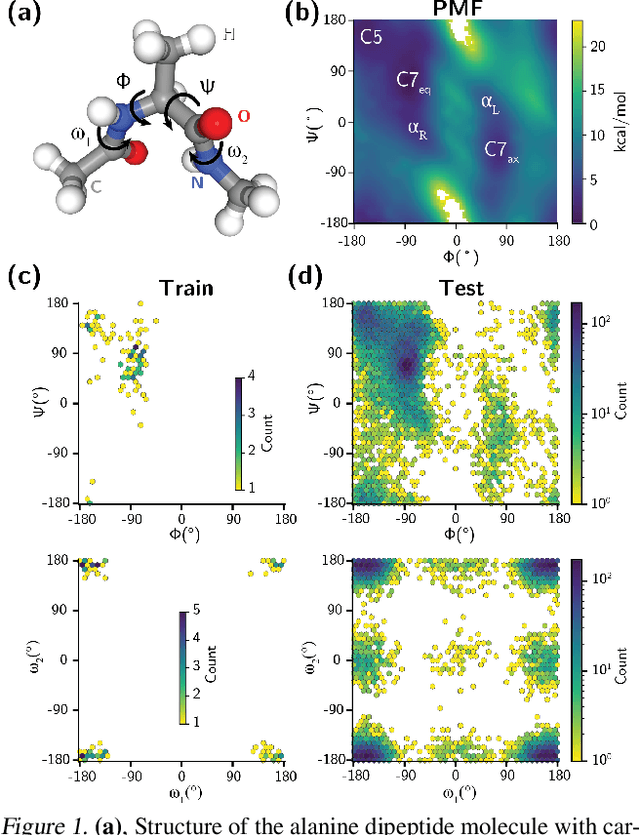
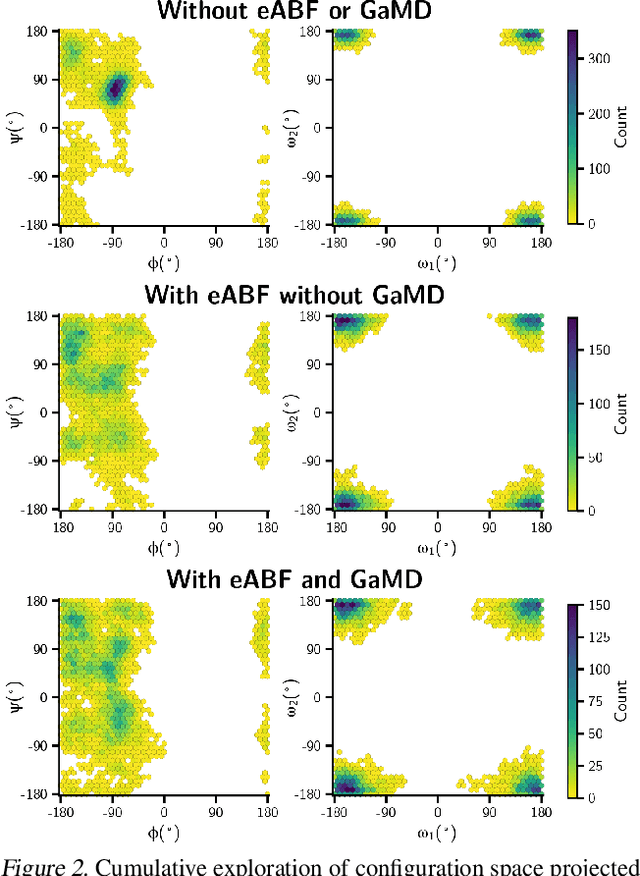
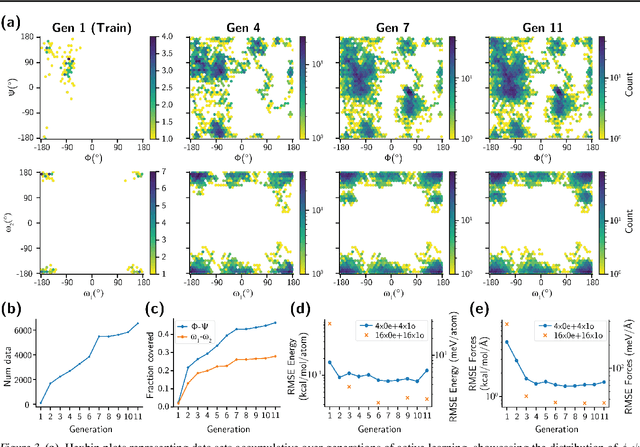
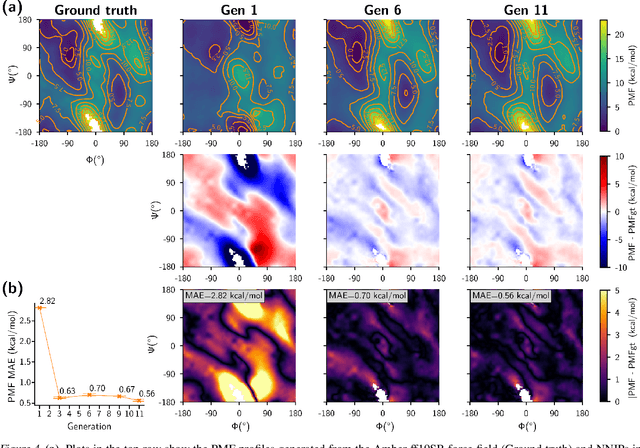
Generating a data set that is representative of the accessible configuration space of a molecular system is crucial for the robustness of machine learned interatomic potentials (MLIP). However, the complexity of molecular systems, characterized by intricate potential energy surfaces (PESs) with numerous local minima and energy barriers, presents a significant challenge. Traditional methods of data generation, such as random sampling or exhaustive exploration, are either intractable or may not capture rare, but highly informative configurations. In this study, we propose a method that leverages uncertainty as the collective variable (CV) to guide the acquisition of chemically-relevant data points, focusing on regions of the configuration space where ML model predictions are most uncertain. This approach employs a Gaussian Mixture Model-based uncertainty metric from a single model as the CV for biased molecular dynamics simulations. The effectiveness of our approach in overcoming energy barriers and exploring unseen energy minima, thereby enhancing the data set in an active learning framework, is demonstrated on the alanine dipeptide benchmark system.
Single-model uncertainty quantification in neural network potentials does not consistently outperform model ensembles
May 02, 2023Neural networks (NNs) often assign high confidence to their predictions, even for points far out-of-distribution, making uncertainty quantification (UQ) a challenge. When they are employed to model interatomic potentials in materials systems, this problem leads to unphysical structures that disrupt simulations, or to biased statistics and dynamics that do not reflect the true physics. Differentiable UQ techniques can find new informative data and drive active learning loops for robust potentials. However, a variety of UQ techniques, including newly developed ones, exist for atomistic simulations and there are no clear guidelines for which are most effective or suitable for a given case. In this work, we examine multiple UQ schemes for improving the robustness of NN interatomic potentials (NNIPs) through active learning. In particular, we compare incumbent ensemble-based methods against strategies that use single, deterministic NNs: mean-variance estimation, deep evidential regression, and Gaussian mixture models. We explore three datasets ranging from in-domain interpolative learning to more extrapolative out-of-domain generalization challenges: rMD17, ammonia inversion, and bulk silica glass. Performance is measured across multiple metrics relating model error to uncertainty. Our experiments show that none of the methods consistently outperformed each other across the various metrics. Ensembling remained better at generalization and for NNIP robustness; MVE only proved effective for in-domain interpolation, while GMM was better out-of-domain; and evidential regression, despite its promise, was not the preferable alternative in any of the cases. More broadly, cost-effective, single deterministic models cannot yet consistently match or outperform ensembling for uncertainty quantification in NNIPs.
Adversarial Attacks on Uncertainty Enable Active Learning for Neural Network Potentials
Feb 01, 2021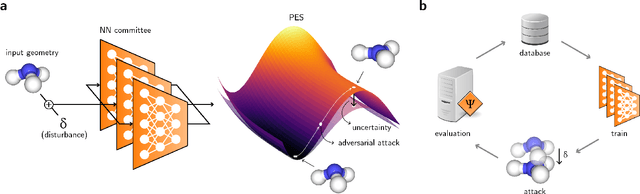
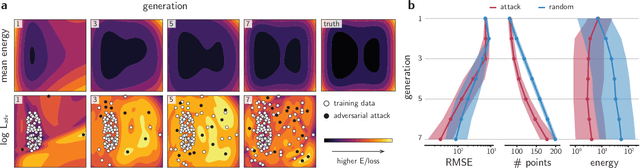
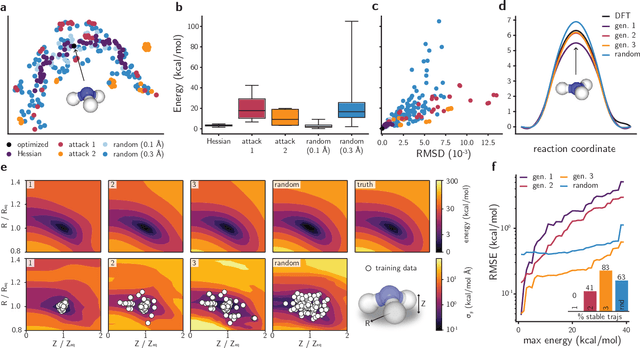
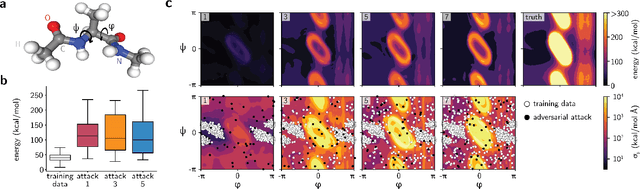
Neural network (NN)-based interatomic potentials provide fast prediction of potential energy surfaces with the accuracy of electronic structure methods. However, NN predictions are only reliable within well-learned training domains, with unknown behavior when extrapolating. Uncertainty quantification through NN committees identify domains with low prediction confidence, but thoroughly exploring the configuration space for training NN potentials often requires slow atomistic simulations. Here, we employ adversarial attacks with a differentiable uncertainty metric to sample new molecular geometries and bootstrap NN potentials. In combination with an active learning loop, the extrapolation power of NN potentials is improved beyond the original training data with few additional samples. The framework is demonstrated on multiple examples, leading to better sampling of kinetic barriers and collective variables without extensive prior data on the relevant geometries. Adversarial attacks are new ways to simultaneously sample the phase space and bootstrap NN potentials, increasing their robustness and enabling a faster, accurate prediction of potential energy landscapes.