 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSelf-Conditioned Denoising for Atomistic Representation Learning
Mar 17, 2026The success of large-scale pretraining in NLP and computer vision has catalyzed growing efforts to develop analogous foundation models for the physical sciences. However, pretraining strategies using atomistic data remain underexplored. To date, large-scale supervised pretraining on DFT force-energy labels has provided the strongest performance gains to downstream property prediction, out-performing existing methods of self-supervised learning (SSL) which remain limited to ground-state geometries, and/or single domains of atomistic data. We address these shortcomings with Self-Conditioned Denoising (SCD), a backbone-agnostic reconstruction objective that utilizes self-embeddings for conditional denoising across any domain of atomistic data, including small molecules, proteins, periodic materials, and 'non-equilibrium' geometries. When controlled for backbone architecture and pretraining dataset, SCD significantly outperforms previous SSL methods on downstream benchmarks and matches or exceeds the performance of supervised force-energy pretraining. We show that a small, fast GNN pretrained by SCD can achieve competitive or superior performance to larger models pretrained on significantly larger labeled or unlabeled datasets, across tasks in multiple domains. Our code is available at: https://github.com/TyJPerez/SelfConditionedDenoisingAtoms
Zatom-1: A Multimodal Flow Foundation Model for 3D Molecules and Materials
Feb 24, 2026General-purpose 3D chemical modeling encompasses molecules and materials, requiring both generative and predictive capabilities. However, most existing AI approaches are optimized for a single domain (molecules or materials) and a single task (generation or prediction), which limits representation sharing and transfer. We introduce Zatom-1, the first foundation model that unifies generative and predictive learning of 3D molecules and materials. Zatom-1 is a Transformer trained with a multimodal flow matching objective that jointly models discrete atom types and continuous 3D geometries. This approach supports scalable pretraining with predictable gains as model capacity increases, while enabling fast and stable sampling. We use joint generative pretraining as a universal initialization for downstream multi-task prediction of properties, energies, and forces. Empirically, Zatom-1 matches or outperforms specialized baselines on both generative and predictive benchmarks, while reducing the generative inference time by more than an order of magnitude. Our experiments demonstrate positive predictive transfer between chemical domains from joint generative pretraining: modeling materials during pretraining improves molecular property prediction accuracy.
Physics-Constrained Flow Matching: Sampling Generative Models with Hard Constraints
Jun 04, 2025



Deep generative models have recently been applied to physical systems governed by partial differential equations (PDEs), offering scalable simulation and uncertainty-aware inference. However, enforcing physical constraints, such as conservation laws (linear and nonlinear) and physical consistencies, remains challenging. Existing methods often rely on soft penalties or architectural biases that fail to guarantee hard constraints. In this work, we propose Physics-Constrained Flow Matching (PCFM), a zero-shot inference framework that enforces arbitrary nonlinear constraints in pretrained flow-based generative models. PCFM continuously guides the sampling process through physics-based corrections applied to intermediate solution states, while remaining aligned with the learned flow and satisfying physical constraints. Empirically, PCFM outperforms both unconstrained and constrained baselines on a range of PDEs, including those with shocks, discontinuities, and sharp features, while ensuring exact constraint satisfaction at the final solution. Our method provides a general framework for enforcing hard constraints in both scientific and general-purpose generative models, especially in applications where constraint satisfaction is essential.
Known Unknowns: Out-of-Distribution Property Prediction in Materials and Molecules
Feb 09, 2025Discovery of high-performance materials and molecules requires identifying extremes with property values that fall outside the known distribution. Therefore, the ability to extrapolate to out-of-distribution (OOD) property values is critical for both solid-state materials and molecular design. Our objective is to train predictor models that extrapolate zero-shot to higher ranges than in the training data, given the chemical compositions of solids or molecular graphs and their property values. We propose using a transductive approach to OOD property prediction, achieving improvements in prediction accuracy. In particular, the True Positive Rate (TPR) of OOD classification of materials and molecules improved by 3x and 2.5x, respectively, and precision improved by 2x and 1.5x compared to non-transductive baselines. Our method leverages analogical input-target relations in the training and test sets, enabling generalization beyond the training target support, and can be applied to any other material and molecular tasks.
Enhanced sampling of robust molecular datasets with uncertainty-based collective variables
Feb 06, 2024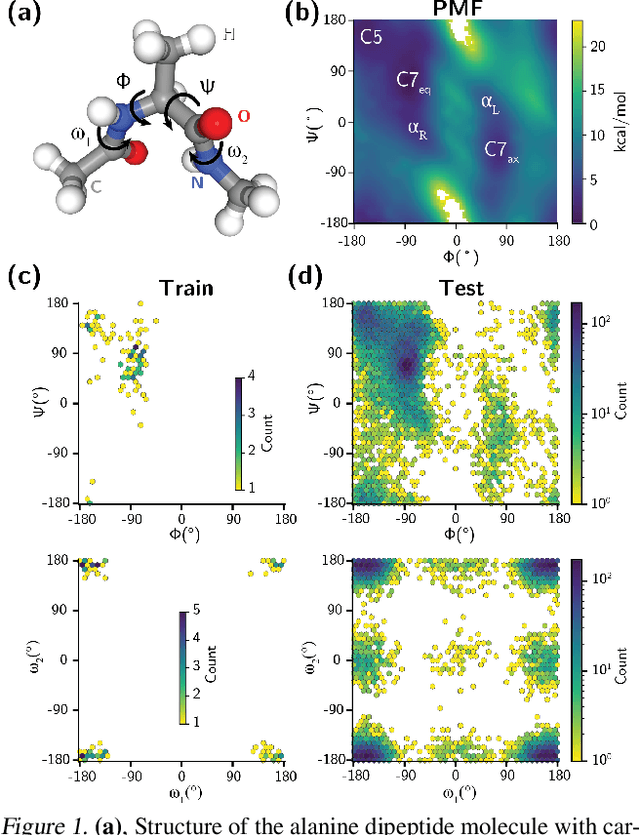
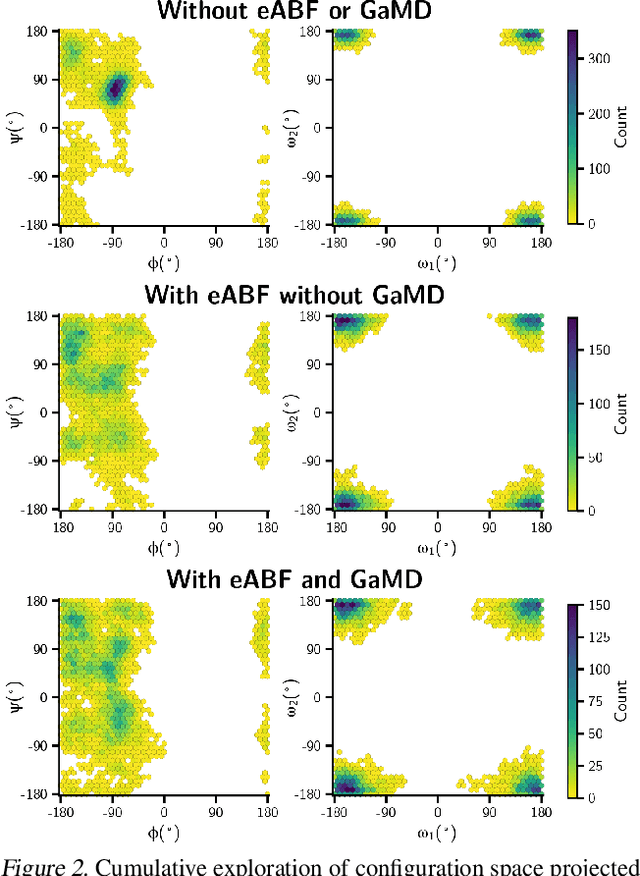
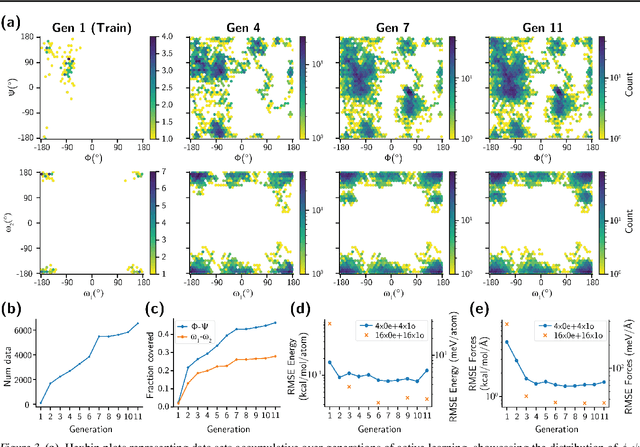
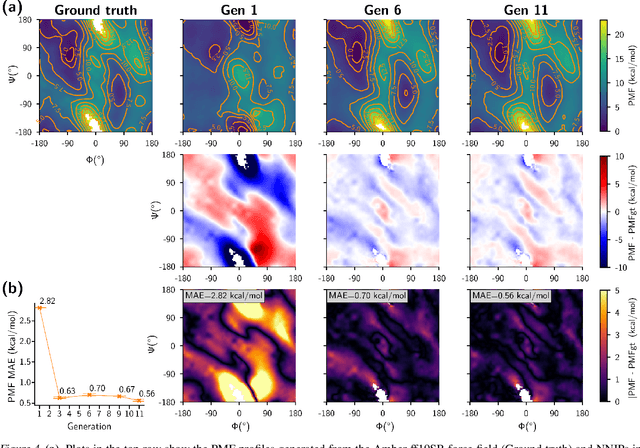
Generating a data set that is representative of the accessible configuration space of a molecular system is crucial for the robustness of machine learned interatomic potentials (MLIP). However, the complexity of molecular systems, characterized by intricate potential energy surfaces (PESs) with numerous local minima and energy barriers, presents a significant challenge. Traditional methods of data generation, such as random sampling or exhaustive exploration, are either intractable or may not capture rare, but highly informative configurations. In this study, we propose a method that leverages uncertainty as the collective variable (CV) to guide the acquisition of chemically-relevant data points, focusing on regions of the configuration space where ML model predictions are most uncertain. This approach employs a Gaussian Mixture Model-based uncertainty metric from a single model as the CV for biased molecular dynamics simulations. The effectiveness of our approach in overcoming energy barriers and exploring unseen energy minima, thereby enhancing the data set in an active learning framework, is demonstrated on the alanine dipeptide benchmark system.
Forces are not Enough: Benchmark and Critical Evaluation for Machine Learning Force Fields with Molecular Simulations
Oct 13, 2022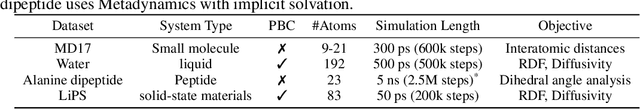
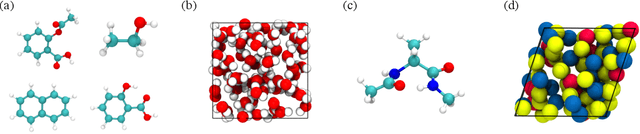

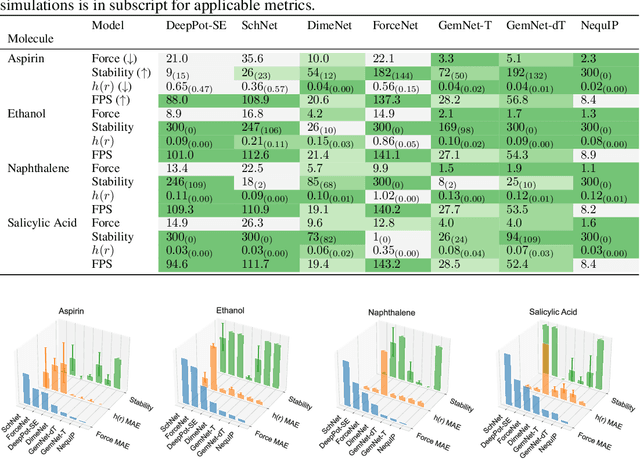
Molecular dynamics (MD) simulation techniques are widely used for various natural science applications. Increasingly, machine learning (ML) force field (FF) models begin to replace ab-initio simulations by predicting forces directly from atomic structures. Despite significant progress in this area, such techniques are primarily benchmarked by their force/energy prediction errors, even though the practical use case would be to produce realistic MD trajectories. We aim to fill this gap by introducing a novel benchmark suite for ML MD simulation. We curate representative MD systems, including water, organic molecules, peptide, and materials, and design evaluation metrics corresponding to the scientific objectives of respective systems. We benchmark a collection of state-of-the-art (SOTA) ML FF models and illustrate, in particular, how the commonly benchmarked force accuracy is not well aligned with relevant simulation metrics. We demonstrate when and how selected SOTA methods fail, along with offering directions for further improvement. Specifically, we identify stability as a key metric for ML models to improve. Our benchmark suite comes with a comprehensive open-source codebase for training and simulation with ML FFs to facilitate further work.
Thermal half-lives of azobenzene derivatives: virtual screening based on intersystem crossing using a machine learning potential
Jul 26, 2022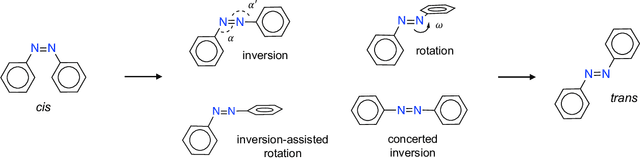
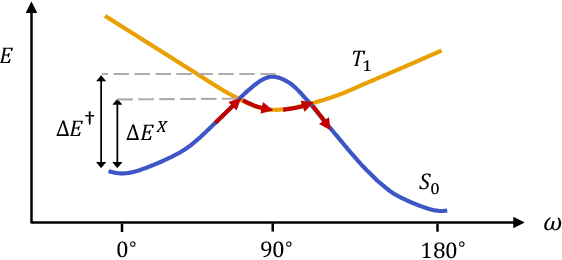
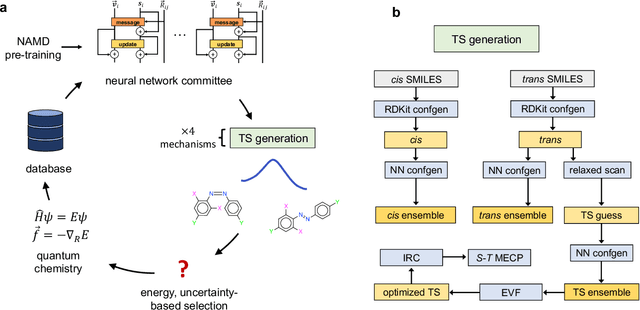
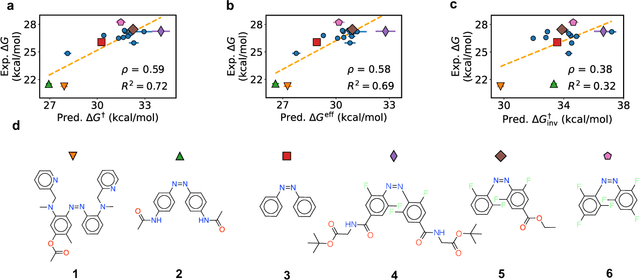
Molecular photoswitches are the foundation of light-activated drugs. A key photoswitch is azobenzene, which exhibits trans-cis isomerism in response to light. The thermal half-life of the cis isomer is of crucial importance, since it controls the duration of the light-induced biological effect. Here we introduce a computational tool for predicting the thermal half-lives of azobenzene derivatives. Our automated approach uses a fast and accurate machine learning potential trained on quantum chemistry data. Building on well-established earlier evidence, we argue that thermal isomerization proceeds through rotation mediated by intersystem crossing, and incorporate this mechanism into our automated workflow. We use our approach to predict the thermal half-lives of 19,000 azobenzene derivatives. We explore trends and tradeoffs between barriers and absorption wavelengths, and open-source our data and software to accelerate research in photopharmacology.
An End-to-End Framework for Molecular Conformation Generation via Bilevel Programming
Jun 02, 2021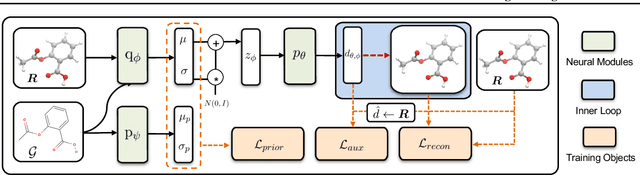
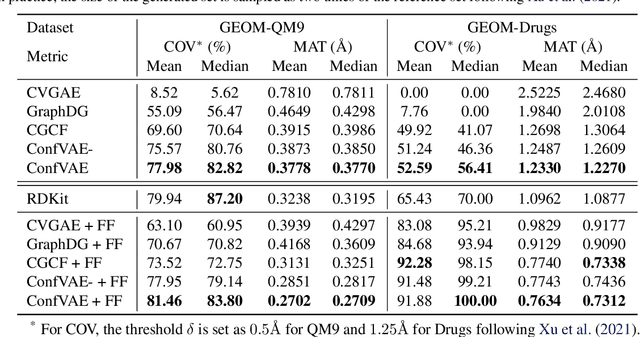
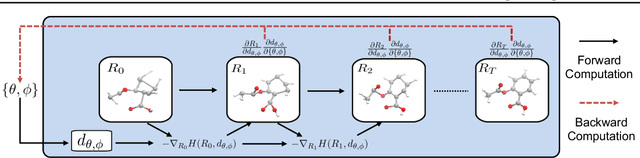
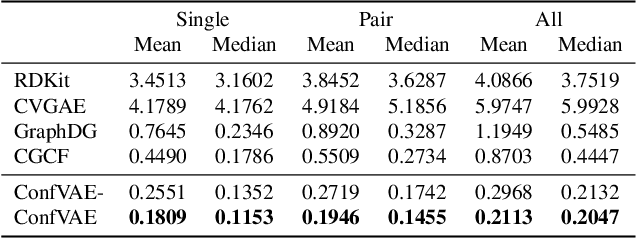
Predicting molecular conformations (or 3D structures) from molecular graphs is a fundamental problem in many applications. Most existing approaches are usually divided into two steps by first predicting the distances between atoms and then generating a 3D structure through optimizing a distance geometry problem. However, the distances predicted with such two-stage approaches may not be able to consistently preserve the geometry of local atomic neighborhoods, making the generated structures unsatisfying. In this paper, we propose an end-to-end solution for molecular conformation prediction called ConfVAE based on the conditional variational autoencoder framework. Specifically, the molecular graph is first encoded in a latent space, and then the 3D structures are generated by solving a principled bilevel optimization program. Extensive experiments on several benchmark data sets prove the effectiveness of our proposed approach over existing state-of-the-art approaches. Code is available at \url{https://github.com/MinkaiXu/ConfVAE-ICML21}.
Accelerating the screening of amorphous polymer electrolytes by learning to reduce random and systematic errors in molecular dynamics simulations
Jan 13, 2021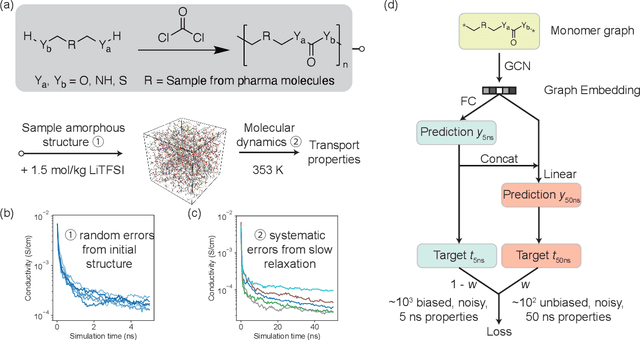
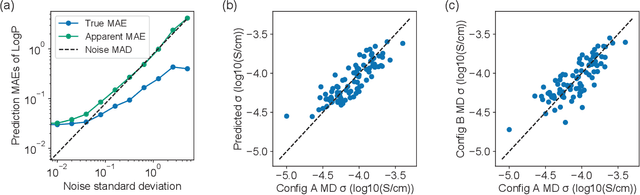
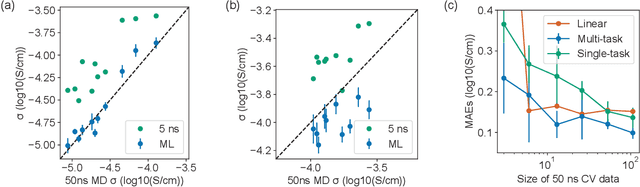
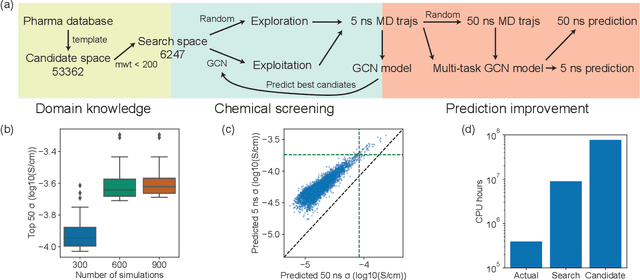
Machine learning has been widely adopted to accelerate the screening of materials. Most existing studies implicitly assume that the training data are generated through a deterministic, unbiased process, but this assumption might not hold for the simulation of some complex materials. In this work, we aim to screen amorphous polymer electrolytes which are promising candidates for the next generation lithium-ion battery technology but extremely expensive to simulate due to their structural complexity. We demonstrate that a multi-task graph neural network can learn from a large amount of noisy, biased data and a small number of unbiased data and reduce both random and systematic errors in predicting the transport properties of polymer electrolytes. This observation allows us to achieve accurate predictions on the properties of complex materials by learning to reduce errors in the training data, instead of running repetitive, expensive simulations which is conventionally used to reduce simulation errors. With this approach, we screen a space of 6247 polymer electrolytes, orders of magnitude larger than previous computational studies. We also find a good extrapolation performance to the top polymers from a larger space of 53362 polymers and 31 experimentally-realized polymers. The strategy employed in this work may be applicable to a broad class of material discovery problems that involve the simulation of complex, amorphous materials.
Molecular machine learning with conformer ensembles
Dec 15, 2020
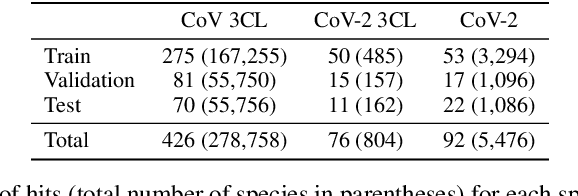
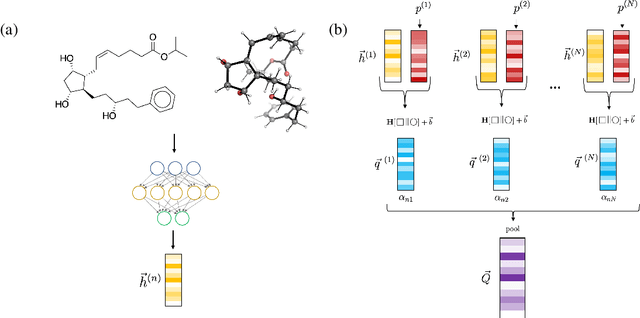
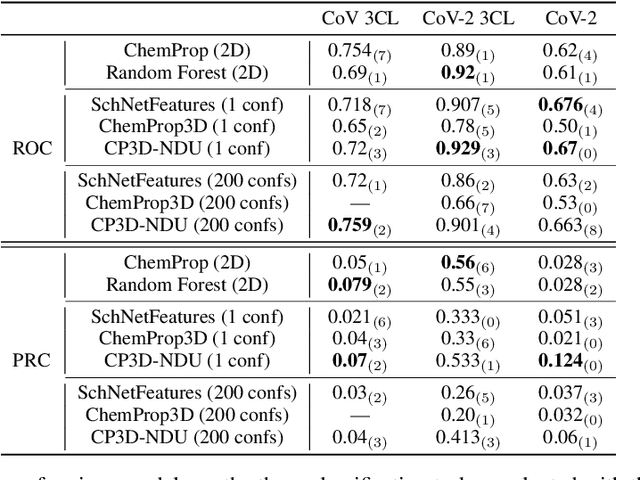
Virtual screening can accelerate drug discovery by identifying top candidates for experimental testing. Machine learning is a powerful method for screening, as it can learn complex structure-property relationships from experimental data and make rapid predictions over virtual libraries. Although molecules are inherently three-dimensional and their biological action typically occurs through supramolecular recognition, most machine learning approaches use a 2D graph representation of molecules as input; few use 3D information, and none take into account the ensemble of conformers accessible to a species. Here we investigate whether the 3D information of multiple conformers can improve molecular property prediction. We introduce a number of new 3D-based models that can take multiple conformers as input to predict drug activity, and find that they learn interpretable weights for each conformer. The new architectures perform significantly better than 2D models, but their performance is just as strong with a single conformer as with many. From this analysis we identify the best 3D architecture and examine its predictions on species without experimental data.