 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolecular machine learning with conformer ensembles
Paper and Code
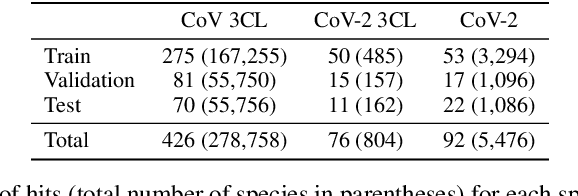
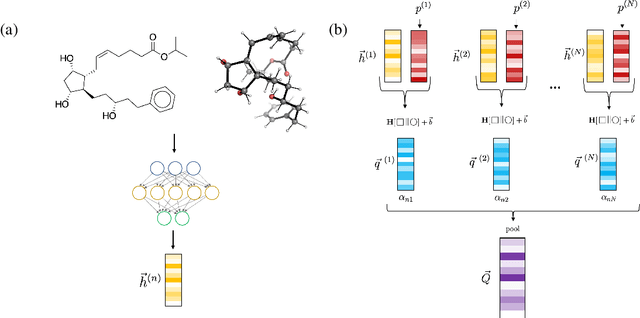
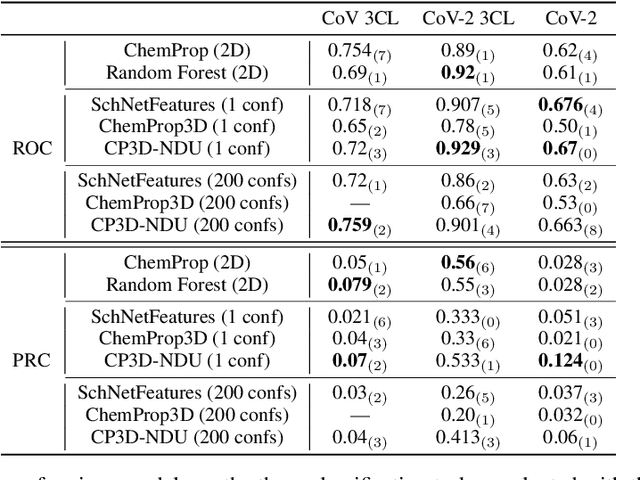
Virtual screening can accelerate drug discovery by identifying top candidates for experimental testing. Machine learning is a powerful method for screening, as it can learn complex structure-property relationships from experimental data and make rapid predictions over virtual libraries. Although molecules are inherently three-dimensional and their biological action typically occurs through supramolecular recognition, most machine learning approaches use a 2D graph representation of molecules as input; few use 3D information, and none take into account the ensemble of conformers accessible to a species. Here we investigate whether the 3D information of multiple conformers can improve molecular property prediction. We introduce a number of new 3D-based models that can take multiple conformers as input to predict drug activity, and find that they learn interpretable weights for each conformer. The new architectures perform significantly better than 2D models, but their performance is just as strong with a single conformer as with many. From this analysis we identify the best 3D architecture and examine its predictions on species without experimental data.