 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGEOM: Energy-annotated molecular conformations for property prediction and molecular generation
Paper and Code
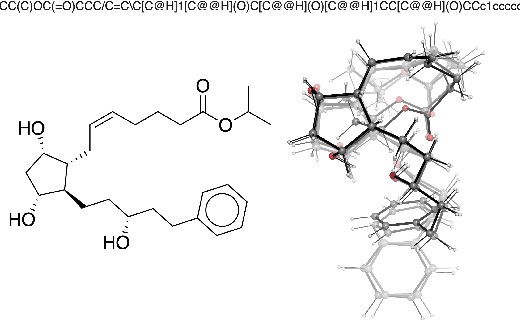
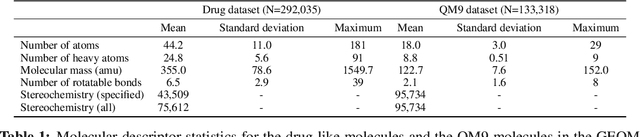


Machine learning outperforms traditional approaches in many molecular design tasks. Although molecules are often thought of as 2D graphs, they in fact consist of an ensemble of inter-converting 3D structures called conformers. Molecular properties arise from the contribution of many conformers, and in the case of a drug binding a target, may be due mainly to a few distinct members. Molecular representations in machine learning are typically based on either one single 3D conformer or on a 2D graph that strips geometrical information. No reference datasets exist that connect these graph and point cloud ensemble representations. Here, we use first-principles simulations to annotate over 400,000 molecules with the ensemble of geometries they span. The Geometrical Embedding Of Molecules (GEOM) dataset contains over 33 million molecular conformers labeled with their relative energies and statistical probabilities at room temperature. This dataset will assist benchmarking and transfer learning in two classes of tasks: inferring 3D properties from 2D molecular graphs, and developing generative models to sample 3D conformations.