 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccelerating the screening of amorphous polymer electrolytes by learning to reduce random and systematic errors in molecular dynamics simulations
Jan 13, 2021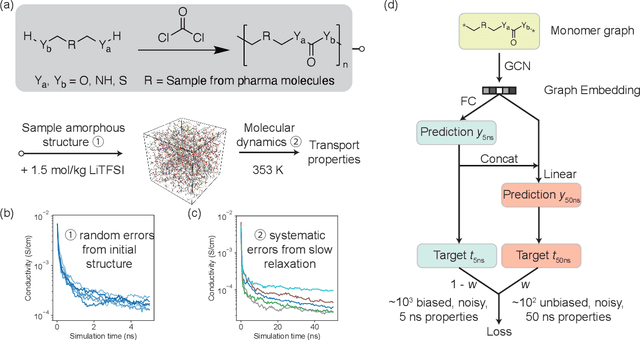
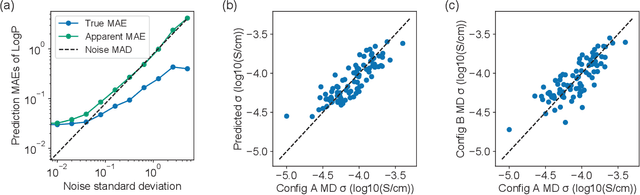
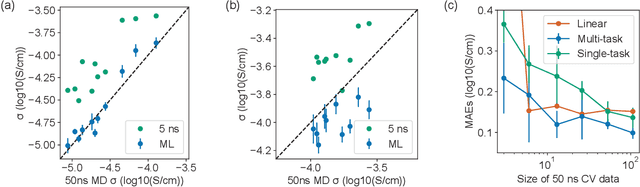
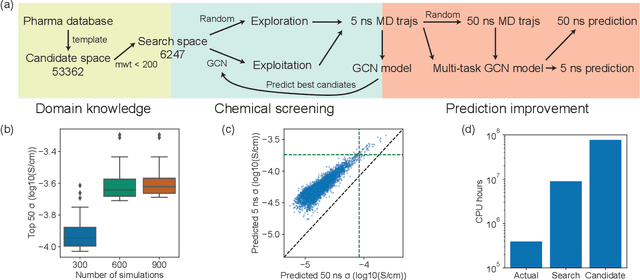
Machine learning has been widely adopted to accelerate the screening of materials. Most existing studies implicitly assume that the training data are generated through a deterministic, unbiased process, but this assumption might not hold for the simulation of some complex materials. In this work, we aim to screen amorphous polymer electrolytes which are promising candidates for the next generation lithium-ion battery technology but extremely expensive to simulate due to their structural complexity. We demonstrate that a multi-task graph neural network can learn from a large amount of noisy, biased data and a small number of unbiased data and reduce both random and systematic errors in predicting the transport properties of polymer electrolytes. This observation allows us to achieve accurate predictions on the properties of complex materials by learning to reduce errors in the training data, instead of running repetitive, expensive simulations which is conventionally used to reduce simulation errors. With this approach, we screen a space of 6247 polymer electrolytes, orders of magnitude larger than previous computational studies. We also find a good extrapolation performance to the top polymers from a larger space of 53362 polymers and 31 experimentally-realized polymers. The strategy employed in this work may be applicable to a broad class of material discovery problems that involve the simulation of complex, amorphous materials.