 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeData-driven path collective variables
Dec 21, 2023



Identifying optimal collective variables to model transformations, using atomic-scale simulations, is a long-standing challenge. We propose a new method for the generation, optimization, and comparison of collective variables, which can be thought of as a data-driven generalization of the path collective variable concept. It consists in a kernel ridge regression of the committor probability, which encodes a transformation's progress. The resulting collective variable is one-dimensional, interpretable, and differentiable, making it appropriate for enhanced sampling simulations requiring biasing. We demonstrate the validity of the method on two different applications: a precipitation model, and the association of Li$^+$ and F$^-$ in water. For the former, we show that global descriptors such as the permutation invariant vector allow to reach an accuracy far from the one achieved \textit{via} simpler, more intuitive variables. For the latter, we show that information correlated with the transformation mechanism is contained in the first solvation shell only, and that inertial effects prevent the derivation of optimal collective variables from the atomic positions only.
A cloud platform for automating and sharing analysis of raw simulation data from high throughput polymer molecular dynamics simulations
Aug 02, 2022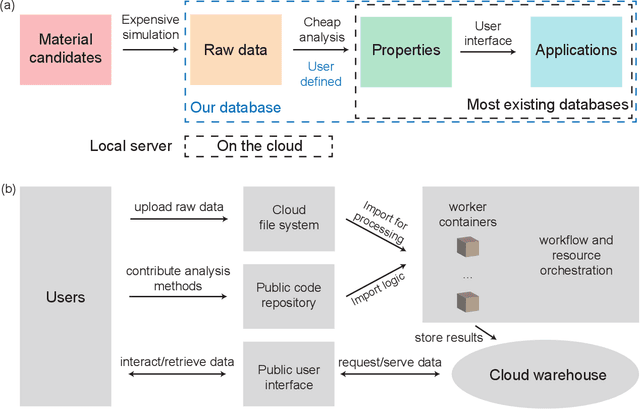
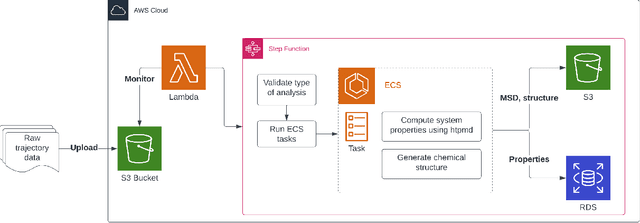
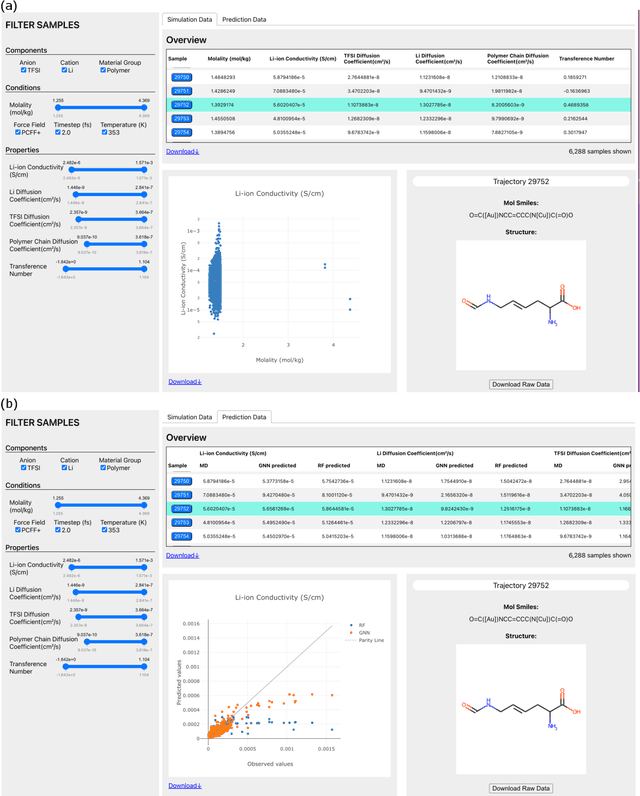
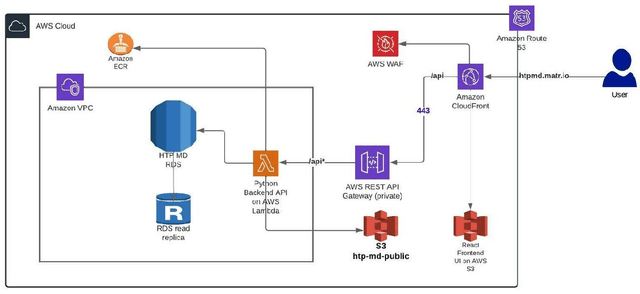
Open material databases storing hundreds of thousands of material structures and their corresponding properties have become the cornerstone of modern computational materials science. Yet, the raw outputs of the simulations, such as the trajectories from molecular dynamics simulations and charge densities from density functional theory calculations, are generally not shared due to their huge size. In this work, we describe a cloud-based platform to facilitate the sharing of raw data and enable the fast post-processing in the cloud to extract new properties defined by the user. As an initial demonstration, our database currently includes 6286 molecular dynamics trajectories for amorphous polymer electrolytes and 5.7 terabytes of data. We create a public analysis library at https://github.com/TRI-AMDD/htp_md to extract multiple properties from the raw data, using both expert designed functions and machine learning models. The analysis is run automatically with computation in the cloud, and results then populate a database that can be accessed publicly. Our platform encourages users to contribute both new trajectory data and analysis functions via public interfaces. Newly analyzed properties will be incorporated into the database. Finally, we create a front-end user interface at https://www.htpmd.matr.io for browsing and visualization of our data. We envision the platform to be a new way of sharing raw data and new insights for the computational materials science community.
Accelerating the screening of amorphous polymer electrolytes by learning to reduce random and systematic errors in molecular dynamics simulations
Jan 13, 2021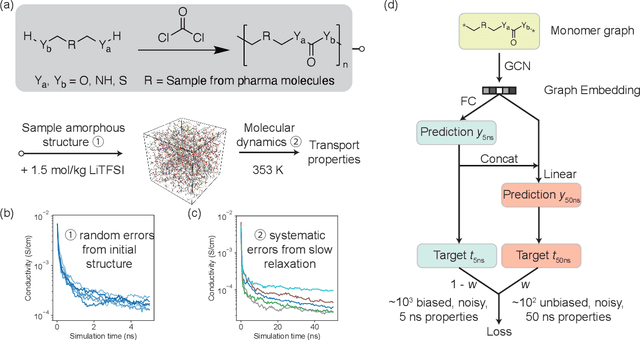
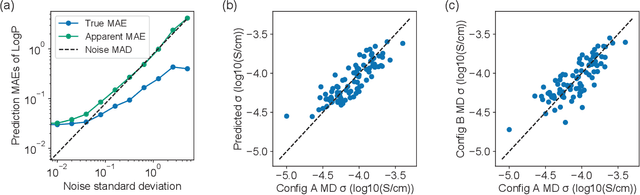
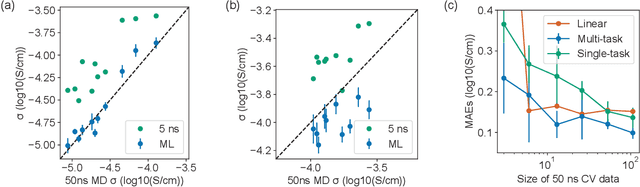
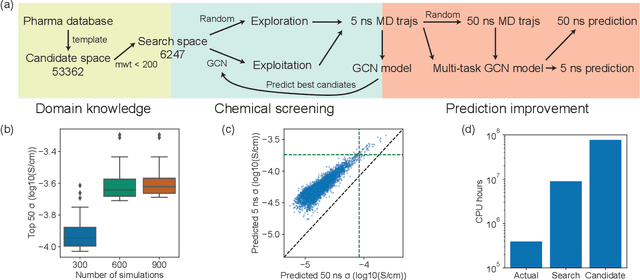
Machine learning has been widely adopted to accelerate the screening of materials. Most existing studies implicitly assume that the training data are generated through a deterministic, unbiased process, but this assumption might not hold for the simulation of some complex materials. In this work, we aim to screen amorphous polymer electrolytes which are promising candidates for the next generation lithium-ion battery technology but extremely expensive to simulate due to their structural complexity. We demonstrate that a multi-task graph neural network can learn from a large amount of noisy, biased data and a small number of unbiased data and reduce both random and systematic errors in predicting the transport properties of polymer electrolytes. This observation allows us to achieve accurate predictions on the properties of complex materials by learning to reduce errors in the training data, instead of running repetitive, expensive simulations which is conventionally used to reduce simulation errors. With this approach, we screen a space of 6247 polymer electrolytes, orders of magnitude larger than previous computational studies. We also find a good extrapolation performance to the top polymers from a larger space of 53362 polymers and 31 experimentally-realized polymers. The strategy employed in this work may be applicable to a broad class of material discovery problems that involve the simulation of complex, amorphous materials.
Graph Dynamical Networks: Unsupervised Learning of Atomic Scale Dynamics in Materials
Feb 18, 2019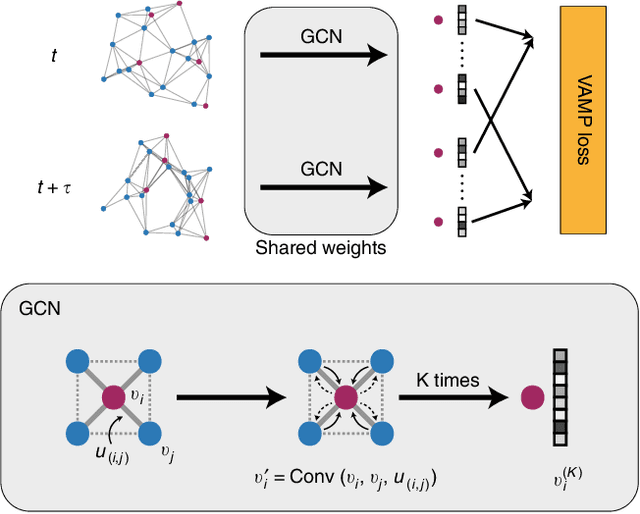
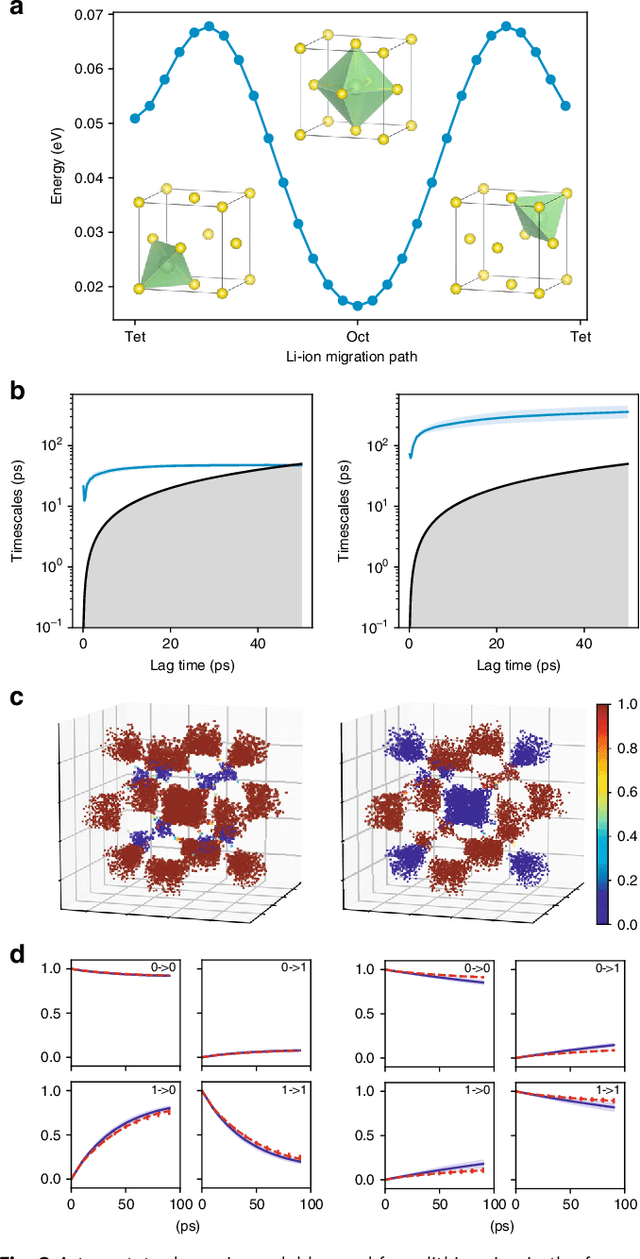
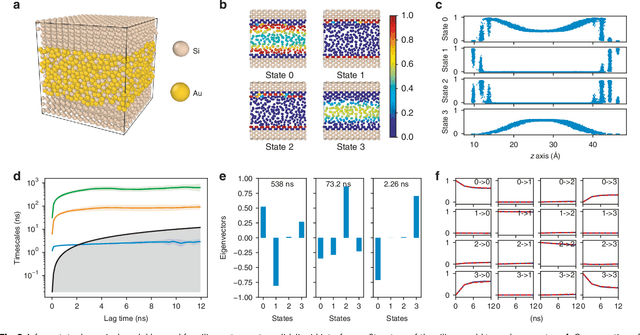
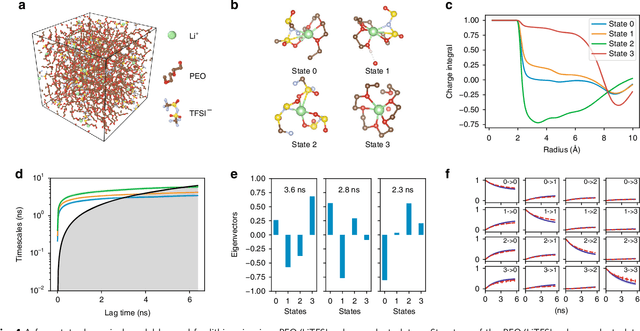
Understanding the dynamical processes that govern the performance of functional materials is essential for the design of next generation materials to tackle global energy and environmental challenges. Many of these processes involve the dynamics of individual atoms or small molecules in condensed phases, e.g. lithium ions in electrolytes, water molecules in membranes, molten atoms at interfaces, etc., which are difficult to understand due to the complexity of local environments. In this work, we develop graph dynamical networks, an unsupervised learning approach for understanding atomic scale dynamics in arbitrary phases and environments from molecular dynamics simulations. We demonstrate that learning the local dynamics of atoms can be significantly easier than the global dynamics of the entire material system using a toy system. We also apply the method to learn the dynamics of two different systems -- silicon atoms at liquid-solid interfaces, and lithium ions in amorphous polymer electrolytes, and show that our approach gains important dynamical information that is otherwise difficult to obtain. With the large amounts of molecular dynamics data generated everyday in nearly every aspect of materials design, this approach provides a broadly useful, automated tool to understand atomic scale dynamics in material systems.