 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePolyCrysDiff: Controllable Generation of Three-Dimensional Computable Polycrystalline Material Structures
Mar 12, 2026The three-dimensional (3D) microstructures of polycrystalline materials exert a critical influence on their mechanical and physical properties. Realistic, controllable construction of these microstructures is a key step toward elucidating structure-property relationships, yet remains a formidable challenge. Herein, we propose PolyCrysDiff, a framework based on conditional latent diffusion that enables the end-to-end generation of computable 3D polycrystalline microstructures. Comprehensive qualitative and quantitative evaluations demonstrate that PolyCrysDiff faithfully reproduces target grain morphologies, orientation distributions, and 3D spatial correlations, while achieving an $R^2$ over 0.972 on grain attributes (e.g., size and sphericity) control, thereby outperforming mainstream approaches such as Markov random field (MRF)- and convolutional neural network (CNN)-based methods. The computability and physical validity of the generated microstructures are verified through a series of crystal plasticity finite element method (CPFEM) simulations. Leveraging PolyCrysDiff's controllable generative capability, we systematically elucidate how grain-level microstructural characteristics affect the mechanical properties of polycrystalline materials. This development is expected to pave a key step toward accelerated, data-driven optimization and design of polycrystalline materials.
Physics Informed Generative AI Enabling Labour Free Segmentation For Microscopy Analysis
Feb 02, 2026Semantic segmentation of microscopy images is a critical task for high-throughput materials characterisation, yet its automation is severely constrained by the prohibitive cost, subjectivity, and scarcity of expert-annotated data. While physics-based simulations offer a scalable alternative to manual labelling, models trained on such data historically fail to generalise due to a significant domain gap, lacking the complex textures, noise patterns, and imaging artefacts inherent to experimental data. This paper introduces a novel framework for labour-free segmentation that successfully bridges this simulation-to-reality gap. Our pipeline leverages phase-field simulations to generate an abundant source of microstructural morphologies with perfect, intrinsically-derived ground-truth masks. We then employ a Cycle-Consistent Generative Adversarial Network (CycleGAN) for unpaired image-to-image translation, transforming the clean simulations into a large-scale dataset of high-fidelity, realistic SEM images. A U-Net model, trained exclusively on this synthetic data, demonstrated remarkable generalisation when deployed on unseen experimental images, achieving a mean Boundary F1-Score of 0.90 and an Intersection over Union (IOU) of 0.88. Comprehensive validation using t-SNE feature-space projection and Shannon entropy analysis confirms that our synthetic images are statistically and featurally indistinguishable from the real data manifold. By completely decoupling model training from manual annotation, our generative framework transforms a data-scarce problem into one of data abundance, providing a robust and fully automated solution to accelerate materials discovery and analysis.
LLMem: Estimating GPU Memory Usage for Fine-Tuning Pre-Trained LLMs
Apr 16, 2024



Fine-tuning pre-trained large language models (LLMs) with limited hardware presents challenges due to GPU memory constraints. Various distributed fine-tuning methods have been proposed to alleviate memory constraints on GPU. However, determining the most effective method for achieving rapid fine-tuning while preventing GPU out-of-memory issues in a given environment remains unclear. To address this challenge, we introduce LLMem, a solution that estimates the GPU memory consumption when applying distributed fine-tuning methods across multiple GPUs and identifies the optimal method. We conduct GPU memory usage estimation prior to fine-tuning, leveraging the fundamental structure of transformer-based decoder models and the memory usage distribution of each method. Experimental results show that LLMem accurately estimates peak GPU memory usage on a single GPU, with error rates of up to 1.6%. Additionally, it shows an average error rate of 3.0% when applying distributed fine-tuning methods to LLMs with more than a billion parameters on multi-GPU setups.
MRGazer: Decoding Eye Gaze Points from Functional Magnetic Resonance Imaging in Individual Space
Nov 27, 2023Eye-tracking research has proven valuable in understanding numerous cognitive functions. Recently, Frey et al. provided an exciting deep learning method for learning eye movements from fMRI data. However, it needed to co-register fMRI into standard space to obtain eyeballs masks, and thus required additional templates and was time consuming. To resolve this issue, in this paper, we propose a framework named MRGazer for predicting eye gaze points from fMRI in individual space. The MRGazer consisted of eyeballs extraction module and a residual network-based eye gaze prediction. Compared to the previous method, the proposed framework skips the fMRI co-registration step, simplifies the processing protocol and achieves end-to-end eye gaze regression. The proposed method achieved superior performance in a variety of eye movement tasks than the co-registration-based method, and delivered objective results within a shorter time (~ 0.02 Seconds for each volume) than prior method (~0.3 Seconds for each volume).
Multi-objective Generative Design of Three-Dimensional Composite Materials
Feb 26, 2023



Composite materials with 3D architectures are desirable in a variety of applications for the capability of tailoring their properties to meet multiple functional requirements. By the arrangement of materials' internal components, structure design is of great significance in tuning the properties of the composites. However, most of the composite structures are proposed by empirical designs following existing patterns. Hindered by the complexity of 3D structures, it is hard to extract customized structures with multiple desired properties from large design space. Here we report a multi-objective driven Wasserstein generative adversarial network (MDWGAN) to implement inverse designs of 3D composite structures according to given geometrical, structural and mechanical requirements. Our framework consists a GAN based network which generates 3D composite structures possessing with similar geometrical and structural features to the target dataset. Besides, multiple objectives are introduced to our framework for the control of mechanical property and isotropy of the composites. Real time calculation of the properties in training iterations is achieved by an accurate surrogate model. We constructed a small and concise dataset to illustrate our framework. With multiple objectives combined by their weight, and the 3D-GAN act as a soft constraint, our framework is proved to be capable of tuning the properties of the generated composites in multiple aspects, while keeping the selected features of different kinds of structures. The feasibility on small dataset and potential scalability on objectives of other properties make our work a novel, effective approach to provide fast, experience free composite structure designs for various functional materials.
Attention module improves both performance and interpretability of 4D fMRI decoding neural network
Oct 03, 2021
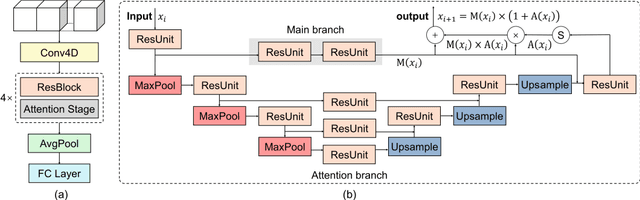

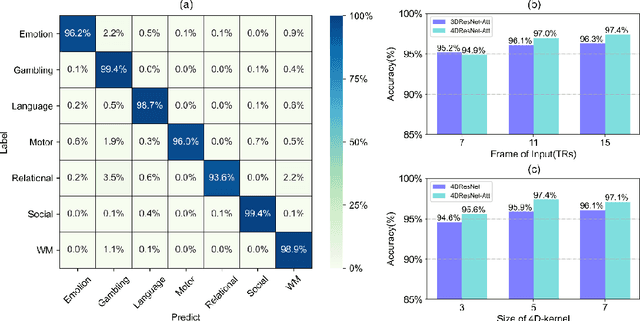
Decoding brain cognitive states from neuroimaging signals is an important topic in neuroscience. In recent years, deep neural networks (DNNs) have been recruited for multiple brain state decoding and achieved good performance. However, the open question of how to interpret the DNN black box remains unanswered. Capitalizing on advances in machine learning, we integrated attention modules into brain decoders to facilitate an in-depth interpretation of DNN channels. A 4D convolution operation was also included to extract temporo-spatial interaction within the fMRI signal. The experiments showed that the proposed model obtains a very high accuracy (97.4%) and outperforms previous researches on the 7 different task benchmarks from the Human Connectome Project (HCP) dataset. The visualization analysis further illustrated the hierarchical emergence of task-specific masks with depth. Finally, the model was retrained to regress individual traits within the HCP and to classify viewing images from the BOLD5000 dataset, respectively. Transfer learning also achieves good performance. A further visualization analysis shows that, after transfer learning, low-level attention masks remained similar to the source domain, whereas high-level attention masks changed adaptively. In conclusion, the proposed 4D model with attention module performed well and facilitated interpretation of DNNs, which is helpful for subsequent research.
Accelerating the screening of amorphous polymer electrolytes by learning to reduce random and systematic errors in molecular dynamics simulations
Jan 13, 2021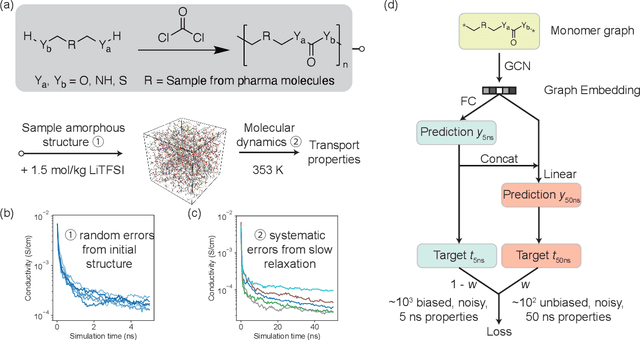
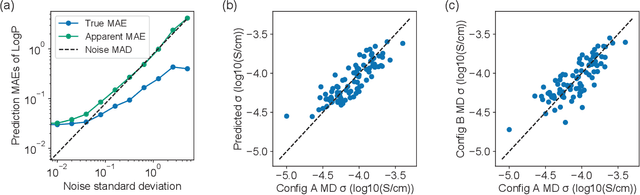
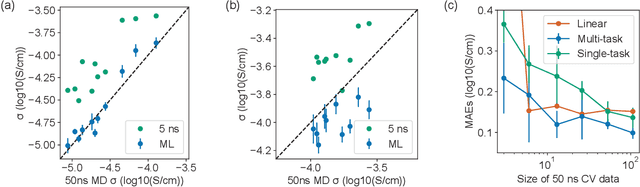
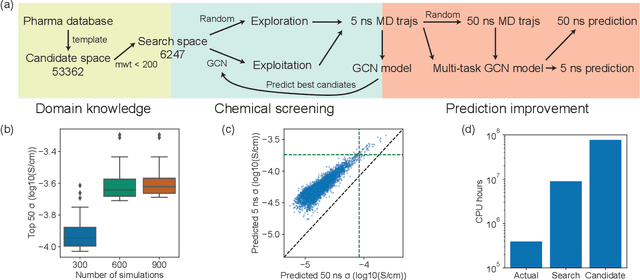
Machine learning has been widely adopted to accelerate the screening of materials. Most existing studies implicitly assume that the training data are generated through a deterministic, unbiased process, but this assumption might not hold for the simulation of some complex materials. In this work, we aim to screen amorphous polymer electrolytes which are promising candidates for the next generation lithium-ion battery technology but extremely expensive to simulate due to their structural complexity. We demonstrate that a multi-task graph neural network can learn from a large amount of noisy, biased data and a small number of unbiased data and reduce both random and systematic errors in predicting the transport properties of polymer electrolytes. This observation allows us to achieve accurate predictions on the properties of complex materials by learning to reduce errors in the training data, instead of running repetitive, expensive simulations which is conventionally used to reduce simulation errors. With this approach, we screen a space of 6247 polymer electrolytes, orders of magnitude larger than previous computational studies. We also find a good extrapolation performance to the top polymers from a larger space of 53362 polymers and 31 experimentally-realized polymers. The strategy employed in this work may be applicable to a broad class of material discovery problems that involve the simulation of complex, amorphous materials.
Searching k-Optimal Goals for an Orienteering Problem on a Specialized Graph with Budget Constraints
Nov 02, 2020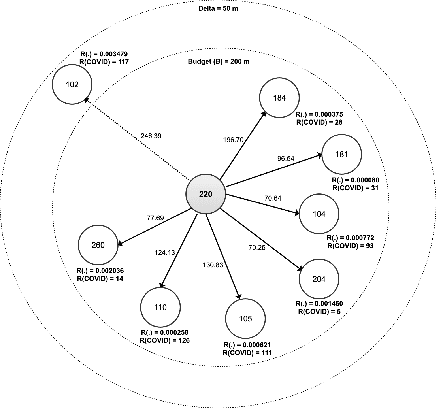
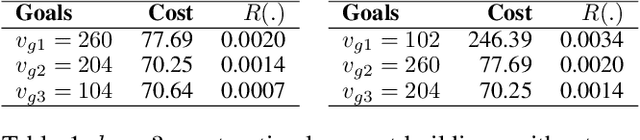

We propose a novel non-randomized anytime orienteering algorithm for finding k-optimal goals that maximize reward on a specialized graph with budget constraints. This specialized graph represents a real-world scenario which is analogous to an orienteering problem of finding k-most optimal goal states.
CHD:Consecutive Horizontal Dropout for Human Gait Feature Extraction
Oct 16, 2019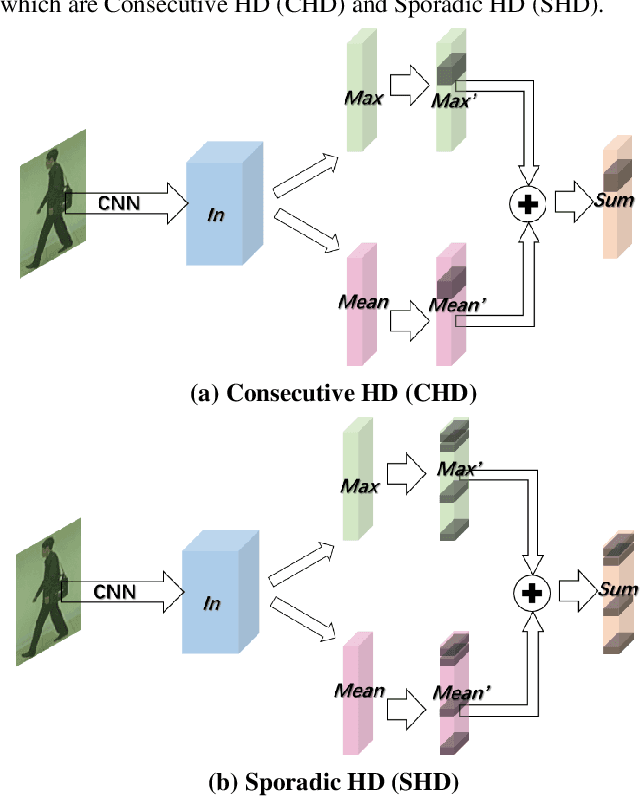
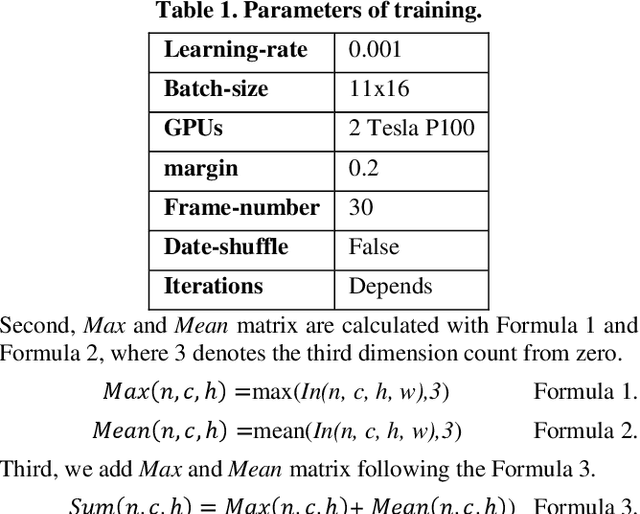
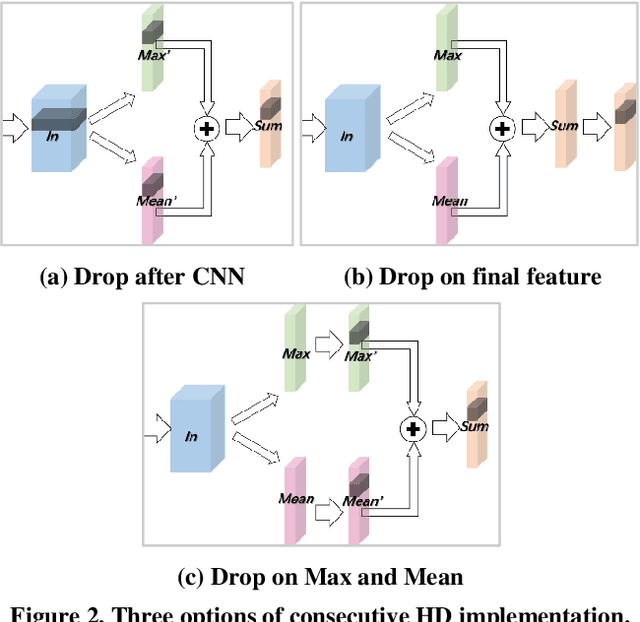
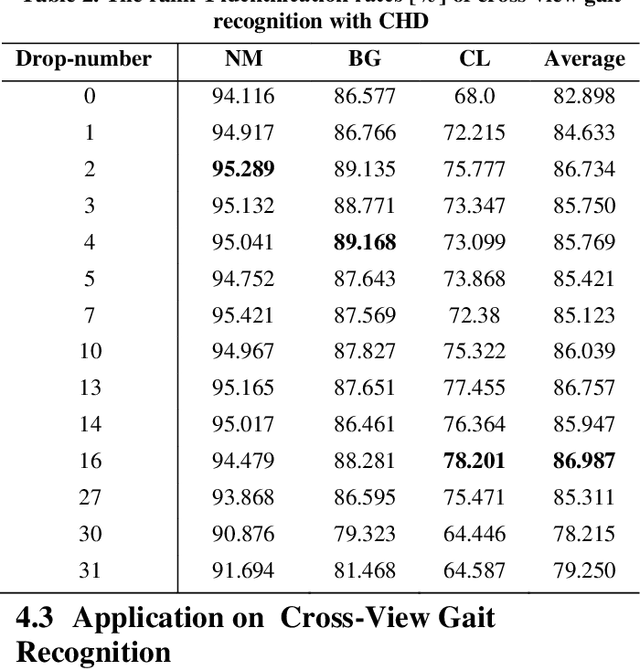
Despite gait recognition and person re-identification researches have made a lot of progress, the accuracy of identification is not high enough in some specific situations, for example, people carrying bags or changing coats. In order to alleviate above situations, we propose a simple but effective Consecutive Horizontal Dropout (CHD) method apply on human feature extraction in deep learning network to avoid overfitting. Within the CHD, we intensify the robust of deep learning network for cross-view gait recognition and person re-identification. The experiments illustrate that the rank-1 accuracy on cross-view gait recognition task has been increased about 10% from 68.0% to 78.201% and 8% from 83.545% to 91.364% in person re-identification task in wearing coat or jacket condition. In addition, 100% accuracy of NM condition was first obtained with CHD. On the benchmarks of CASIA-B, above accuracies are state-of-the-arts.
Graph Dynamical Networks: Unsupervised Learning of Atomic Scale Dynamics in Materials
Feb 18, 2019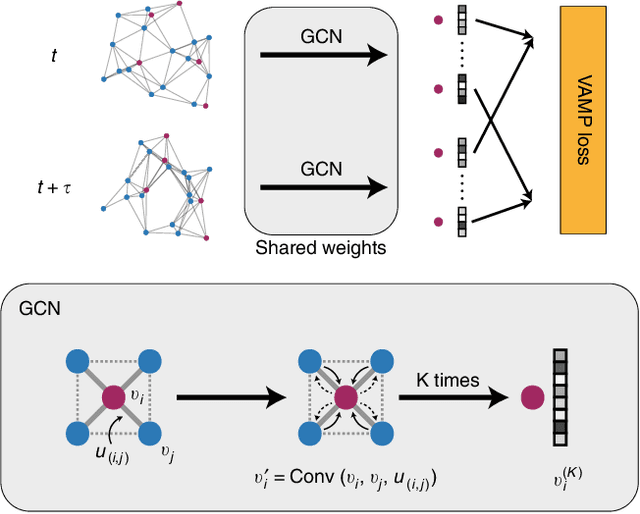
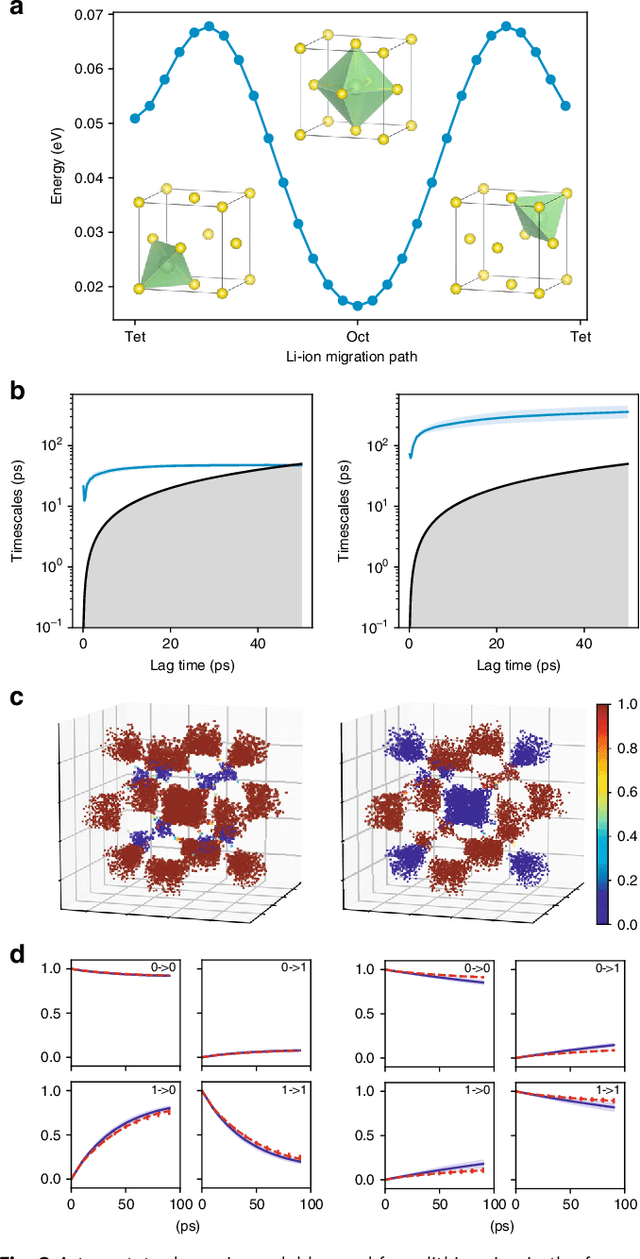
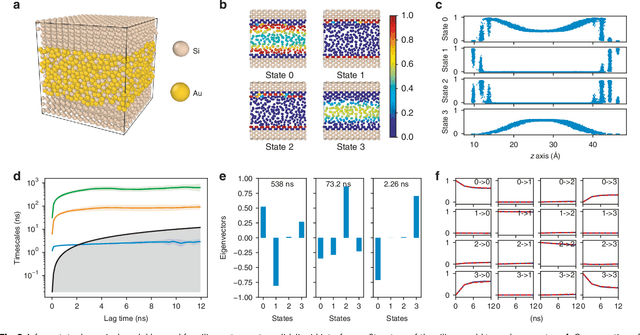
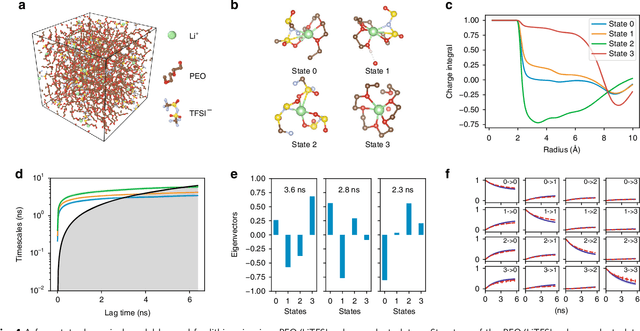
Understanding the dynamical processes that govern the performance of functional materials is essential for the design of next generation materials to tackle global energy and environmental challenges. Many of these processes involve the dynamics of individual atoms or small molecules in condensed phases, e.g. lithium ions in electrolytes, water molecules in membranes, molten atoms at interfaces, etc., which are difficult to understand due to the complexity of local environments. In this work, we develop graph dynamical networks, an unsupervised learning approach for understanding atomic scale dynamics in arbitrary phases and environments from molecular dynamics simulations. We demonstrate that learning the local dynamics of atoms can be significantly easier than the global dynamics of the entire material system using a toy system. We also apply the method to learn the dynamics of two different systems -- silicon atoms at liquid-solid interfaces, and lithium ions in amorphous polymer electrolytes, and show that our approach gains important dynamical information that is otherwise difficult to obtain. With the large amounts of molecular dynamics data generated everyday in nearly every aspect of materials design, this approach provides a broadly useful, automated tool to understand atomic scale dynamics in material systems.