 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeData-Driven Parametrization of Molecular Mechanics Force Fields for Expansive Chemical Space Coverage
Aug 23, 2024



A force field is a critical component in molecular dynamics simulations for computational drug discovery. It must achieve high accuracy within the constraints of molecular mechanics' (MM) limited functional forms, which offers high computational efficiency. With the rapid expansion of synthetically accessible chemical space, traditional look-up table approaches face significant challenges. In this study, we address this issue using a modern data-driven approach, developing ByteFF, an Amber-compatible force field for drug-like molecules. To create ByteFF, we generated an expansive and highly diverse molecular dataset at the B3LYP-D3(BJ)/DZVP level of theory. This dataset includes 2.4 million optimized molecular fragment geometries with analytical Hessian matrices, along with 3.2 million torsion profiles. We then trained an edge-augmented, symmetry-preserving molecular graph neural network (GNN) on this dataset, employing a carefully optimized training strategy. Our model predicts all bonded and non-bonded MM force field parameters for drug-like molecules simultaneously across a broad chemical space. ByteFF demonstrates state-of-the-art performance on various benchmark datasets, excelling in predicting relaxed geometries, torsional energy profiles, and conformational energies and forces. Its exceptional accuracy and expansive chemical space coverage make ByteFF a valuable tool for multiple stages of computational drug discovery.
Crystals with Transformers on Graphs, for Prediction of Unconventional Crystal Material Properties and the Benchmark
Jul 23, 2024The ionic bonding across the lattice and ordered microscopic structures endow crystals with unique symmetry and determine their macroscopic properties. Unconventional crystals, in particular, exhibit non-traditional lattice structures or possess exotic physical properties, making them intriguing subjects for investigation. Therefore, to accurately predict the physical and chemical properties of crystals, it is crucial to consider long-range orders. While GNN excels at capturing the local environment of atoms in crystals, they often face challenges in effectively capturing longer-ranged interactions due to their limited depth. In this paper, we propose CrysToGraph ($\textbf{Crys}$tals with $\textbf{T}$ransformers $\textbf{o}$n $\textbf{Graph}$s), a novel transformer-based geometric graph network designed specifically for unconventional crystalline systems, and UnconvBench, a comprehensive benchmark to evaluate models' predictive performance on unconventional crystal materials such as defected crystals, low-dimension crystals and MOF. CrysToGraph effectively captures short-range interactions with transformer-based graph convolution blocks as well as long-range interactions with graph-wise transformer blocks. CrysToGraph proofs its effectiveness in modelling unconventional crystal materials in multiple tasks, and moreover, it outperforms most existing methods, achieving new state-of-the-art results on the benchmarks of both unconventional crystals and traditional crystals.
BAMBOO: a predictive and transferable machine learning force field framework for liquid electrolyte development
Apr 12, 2024Despite the widespread applications of machine learning force field (MLFF) on solids and small molecules, there is a notable gap in applying MLFF to complex liquid electrolytes. In this work, we introduce BAMBOO (ByteDance AI Molecular Simulation Booster), a novel framework for molecular dynamics (MD) simulations, with a demonstration of its capabilities in the context of liquid electrolytes for lithium batteries. We design a physics-inspired graph equivariant transformer architecture as the backbone of BAMBOO to learn from quantum mechanical simulations. Additionally, we pioneer an ensemble knowledge distillation approach and apply it on MLFFs to improve the stability of MD simulations. Finally, we propose the density alignment algorithm to align BAMBOO with experimental measurements. BAMBOO demonstrates state-of-the-art accuracy in predicting key electrolyte properties such as density, viscosity, and ionic conductivity across various solvents and salt combinations. Our current model, trained on more than 15 chemical species, achieves the average density error of 0.01 g/cm$^3$ on various compositions compared with experimental data. Moreover, our model demonstrates transferability to molecules not included in the quantum mechanical dataset. We envision this work as paving the way to a "universal MLFF" capable of simulating properties of common organic liquids.
A cloud platform for automating and sharing analysis of raw simulation data from high throughput polymer molecular dynamics simulations
Aug 02, 2022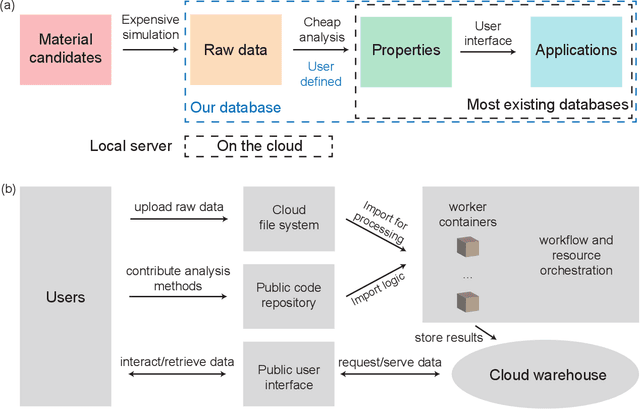
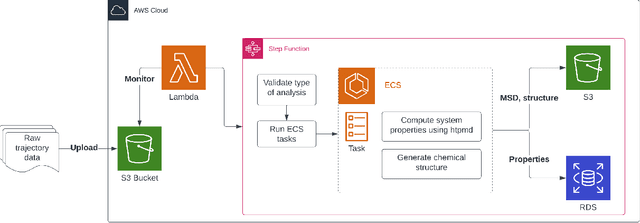
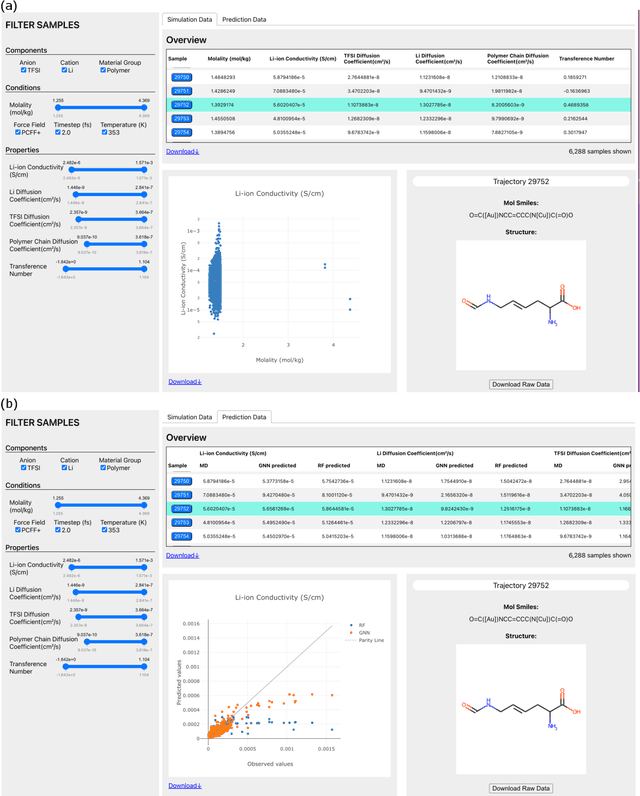
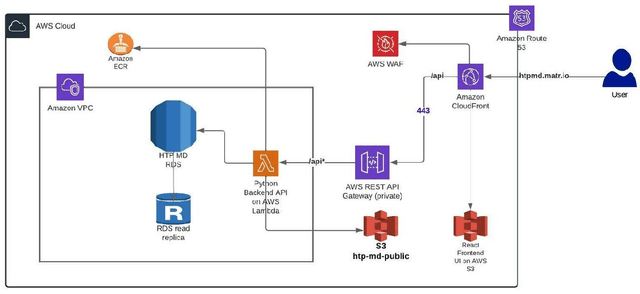
Open material databases storing hundreds of thousands of material structures and their corresponding properties have become the cornerstone of modern computational materials science. Yet, the raw outputs of the simulations, such as the trajectories from molecular dynamics simulations and charge densities from density functional theory calculations, are generally not shared due to their huge size. In this work, we describe a cloud-based platform to facilitate the sharing of raw data and enable the fast post-processing in the cloud to extract new properties defined by the user. As an initial demonstration, our database currently includes 6286 molecular dynamics trajectories for amorphous polymer electrolytes and 5.7 terabytes of data. We create a public analysis library at https://github.com/TRI-AMDD/htp_md to extract multiple properties from the raw data, using both expert designed functions and machine learning models. The analysis is run automatically with computation in the cloud, and results then populate a database that can be accessed publicly. Our platform encourages users to contribute both new trajectory data and analysis functions via public interfaces. Newly analyzed properties will be incorporated into the database. Finally, we create a front-end user interface at https://www.htpmd.matr.io for browsing and visualization of our data. We envision the platform to be a new way of sharing raw data and new insights for the computational materials science community.