 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeData-Driven Parametrization of Molecular Mechanics Force Fields for Expansive Chemical Space Coverage
Aug 23, 2024



A force field is a critical component in molecular dynamics simulations for computational drug discovery. It must achieve high accuracy within the constraints of molecular mechanics' (MM) limited functional forms, which offers high computational efficiency. With the rapid expansion of synthetically accessible chemical space, traditional look-up table approaches face significant challenges. In this study, we address this issue using a modern data-driven approach, developing ByteFF, an Amber-compatible force field for drug-like molecules. To create ByteFF, we generated an expansive and highly diverse molecular dataset at the B3LYP-D3(BJ)/DZVP level of theory. This dataset includes 2.4 million optimized molecular fragment geometries with analytical Hessian matrices, along with 3.2 million torsion profiles. We then trained an edge-augmented, symmetry-preserving molecular graph neural network (GNN) on this dataset, employing a carefully optimized training strategy. Our model predicts all bonded and non-bonded MM force field parameters for drug-like molecules simultaneously across a broad chemical space. ByteFF demonstrates state-of-the-art performance on various benchmark datasets, excelling in predicting relaxed geometries, torsional energy profiles, and conformational energies and forces. Its exceptional accuracy and expansive chemical space coverage make ByteFF a valuable tool for multiple stages of computational drug discovery.
BAMBOO: a predictive and transferable machine learning force field framework for liquid electrolyte development
Apr 12, 2024Despite the widespread applications of machine learning force field (MLFF) on solids and small molecules, there is a notable gap in applying MLFF to complex liquid electrolytes. In this work, we introduce BAMBOO (ByteDance AI Molecular Simulation Booster), a novel framework for molecular dynamics (MD) simulations, with a demonstration of its capabilities in the context of liquid electrolytes for lithium batteries. We design a physics-inspired graph equivariant transformer architecture as the backbone of BAMBOO to learn from quantum mechanical simulations. Additionally, we pioneer an ensemble knowledge distillation approach and apply it on MLFFs to improve the stability of MD simulations. Finally, we propose the density alignment algorithm to align BAMBOO with experimental measurements. BAMBOO demonstrates state-of-the-art accuracy in predicting key electrolyte properties such as density, viscosity, and ionic conductivity across various solvents and salt combinations. Our current model, trained on more than 15 chemical species, achieves the average density error of 0.01 g/cm$^3$ on various compositions compared with experimental data. Moreover, our model demonstrates transferability to molecules not included in the quantum mechanical dataset. We envision this work as paving the way to a "universal MLFF" capable of simulating properties of common organic liquids.
Learning Large-Time-Step Molecular Dynamics with Graph Neural Networks
Dec 21, 2021

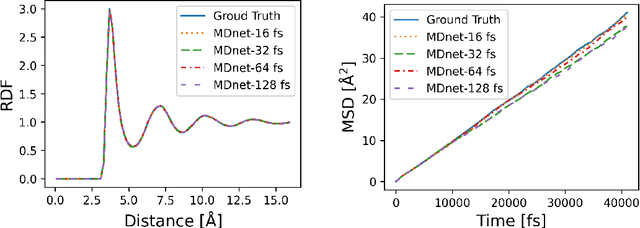
Molecular dynamics (MD) simulation predicts the trajectory of atoms by solving Newton's equation of motion with a numeric integrator. Due to physical constraints, the time step of the integrator need to be small to maintain sufficient precision. This limits the efficiency of simulation. To this end, we introduce a graph neural network (GNN) based model, MDNet, to predict the evolution of coordinates and momentum with large time steps. In addition, MDNet can easily scale to a larger system, due to its linear complexity with respect to the system size. We demonstrate the performance of MDNet on a 4000-atom system with large time steps, and show that MDNet can predict good equilibrium and transport properties, well aligned with standard MD simulations.