 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnabling Real-Time Point-of-Care Ultrasound Segmentation: A GPU-Free Deployment in Resource-Limited Settings
Jun 13, 2026Ultrasound imaging is the most widely adopted medical modality globally due to its low cost and portability, yet artificial intelligence (AI) deployment remains constrained by reliance on GPU-accelerated models, creating a structural paradox where the cost of "intelligence" exceeds that of the imaging device itself. Here, we present the systematic adaptation and extensive evaluation of UltraSeg, an ultra-lightweight architecture originally developed for colonoscopic polyp segmentation, now engineered for point-of-care ultrasound (POCUS) across ten public datasets spanning six anatomical sites (breast, thyroid, kidney, carotid, fetal, and small-animal tumor). We systematically validate both variants in ultrasound domains: UltraSeg-130K (0.13M parameters) achieves 89.7 FPS on single-core CPUs and 34.8 FPS on a refurbished mobile device, while UltraSeg-500K (0.5M parameters) delivers 44.6 FPS on CPU and 16.1 FPS on mobile device. UltraSeg-500K matches or exceeds the Dice performance of the 31M-parameter UNet and approaches 105M-parameter TransUNet in average performance, with superior zero-shot cross-dataset generalization on external validation sets (UDIAT, DDTI). By enabling clinical-grade segmentation without GPU dependency, this work brings AI costs in line with ultrasound accessibility, making advanced diagnostics available in resource-limited settings.
EyeMVP: OCT-Informed Fundus Representation Learning via Paired CFP--OCT Pretraining
Jun 13, 2026Color fundus photography (CFP) is the mainstay for large-scale retinal screening, yet its diagnostic capacity is constrained by the lack of depth-resolved structural information. Optical coherence tomography (OCT) provides cross-sectional retinal anatomy, but is less accessible in population-level screening. Here, we present EyeMVP, a cross-modal retinal foundation model that uses paired CFP--OCT pretraining to learn OCT-informed CFP representations. EyeMVP is pretrained on 674,893 strict same-eye same-day paired CFP--OCT image triples from 112,642 patients across eight hospitals in China. The model uses cross-modal masked reconstruction to enrich CFP representations with OCT-associated supervision, while requiring only CFP images at inference. To accommodate the non-aligned imaging geometry between en-face CFP and cross-sectional OCT, EyeMVP combines source-constrained cross-attention with CFP-derived structural masks. Across 16 downstream tasks, including classification, segmentation, few-shot adaptation, and cross-modal retrieval, EyeMVP outperforms representative retinal foundation models and shows consistent gains on tasks involving macular and optic nerve structure. For CFP-challenging macular diseases, EyeMVP achieves an AUROC of 0.948 for macular edema (vs.~0.852 for EyeCLIP) and 0.825 for myopic macular schisis. In an exploratory reader study, EyeMVP exceeds junior and intermediate ophthalmologist groups but does not reach senior ophthalmologist performance on macular edema, while showing numerically higher balanced accuracy than all reader groups on myopic macular schisis. These results suggest that pixel-level cross-modal reconstruction can enrich CFP representations with OCT-associated supervision, providing a practical route toward stronger CFP-based retinal analysis in screening settings.
Enabling Real-Time Colonoscopic Polyp Segmentation on Commodity CPUs via Ultra-Lightweight Architecture
Feb 04, 2026Early detection of colorectal cancer hinges on real-time, accurate polyp identification and resection. Yet current high-precision segmentation models rely on GPUs, making them impractical to deploy in primary hospitals, mobile endoscopy units, or capsule robots. To bridge this gap, we present the UltraSeg family, operating in an extreme-compression regime (<0.3 M parameters). UltraSeg-108K (0.108 M parameters) is optimized for single-center data, while UltraSeg-130K (0.13 M parameters) generalizes to multi-center, multi-modal images. By jointly optimizing encoder-decoder widths, incorporating constrained dilated convolutions to enlarge receptive fields, and integrating a cross-layer lightweight fusion module, the models achieve 90 FPS on a single CPU core without sacrificing accuracy. Evaluated on seven public datasets, UltraSeg retains >94% of the Dice score of a 31 M-parameter U-Net while utilizing only 0.4% of its parameters, establishing a strong, clinically viable baseline for the extreme-compression domain and offering an immediately deployable solution for resource-constrained settings. This work provides not only a CPU-native solution for colonoscopy but also a reproducible blueprint for broader minimally invasive surgical vision applications. Source code is publicly available to ensure reproducibility and facilitate future benchmarking.
Seed-Prover 1.5: Mastering Undergraduate-Level Theorem Proving via Learning from Experience
Dec 19, 2025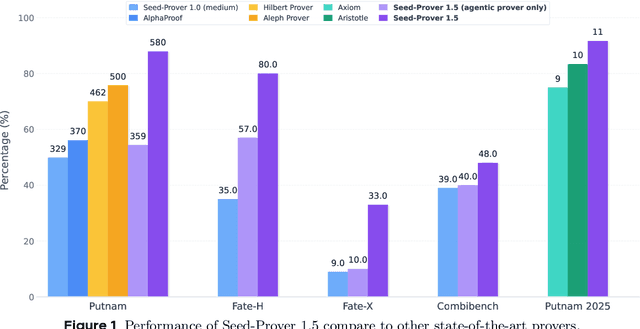

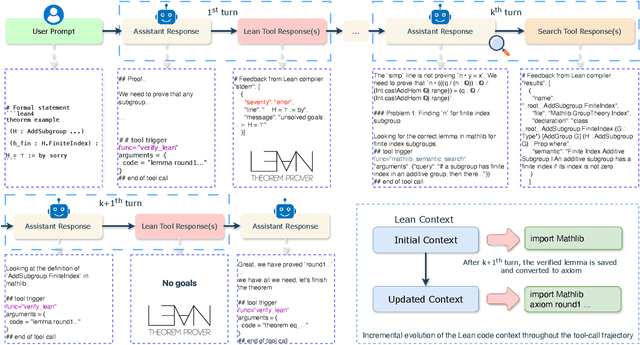

Large language models have recently made significant progress to generate rigorous mathematical proofs. In contrast, utilizing LLMs for theorem proving in formal languages (such as Lean) remains challenging and computationally expensive, particularly when addressing problems at the undergraduate level and beyond. In this work, we present \textbf{Seed-Prover 1.5}, a formal theorem-proving model trained via large-scale agentic reinforcement learning, alongside an efficient test-time scaling (TTS) workflow. Through extensive interactions with Lean and other tools, the model continuously accumulates experience during the RL process, substantially enhancing the capability and efficiency of formal theorem proving. Furthermore, leveraging recent advancements in natural language proving, our TTS workflow efficiently bridges the gap between natural and formal languages. Compared to state-of-the-art methods, Seed-Prover 1.5 achieves superior performance with a smaller compute budget. It solves \textbf{88\% of PutnamBench} (undergraduate-level), \textbf{80\% of Fate-H} (graduate-level), and \textbf{33\% of Fate-X} (PhD-level) problems. Notably, using our system, we solved \textbf{11 out of 12 problems} from Putnam 2025 within 9 hours. Our findings suggest that scaling learning from experience, driven by high-quality formal feedback, holds immense potential for the future of formal mathematical reasoning.
MolSculpt: Sculpting 3D Molecular Geometries from Chemical Syntax
Dec 09, 2025Generating precise 3D molecular geometries is crucial for drug discovery and material science. While prior efforts leverage 1D representations like SELFIES to ensure molecular validity, they fail to fully exploit the rich chemical knowledge entangled within 1D models, leading to a disconnect between 1D syntactic generation and 3D geometric realization. To bridge this gap, we propose MolSculpt, a novel framework that "sculpts" 3D molecular geometries from chemical syntax. MolSculpt is built upon a frozen 1D molecular foundation model and a 3D molecular diffusion model. We introduce a set of learnable queries to extract inherent chemical knowledge from the foundation model, and a trainable projector then injects this cross-modal information into the conditioning space of the diffusion model to guide the 3D geometry generation. In this way, our model deeply integrates 1D latent chemical knowledge into the 3D generation process through end-to-end optimization. Experiments demonstrate that MolSculpt achieves state-of-the-art (SOTA) performance in \textit{de novo} 3D molecule generation and conditional 3D molecule generation, showing superior 3D fidelity and stability on both the GEOM-DRUGS and QM9 datasets. Code is available at https://github.com/SakuraTroyChen/MolSculpt.
A New Deep-learning-Based Approach For mRNA Optimization: High Fidelity, Computation Efficiency, and Multiple Optimization Factors
May 29, 2025The mRNA optimization is critical for therapeutic and biotechnological applications, since sequence features directly govern protein expression levels and efficacy. However, current methods face significant challenges in simultaneously achieving three key objectives: (1) fidelity (preventing unintended amino acid changes), (2) computational efficiency (speed and scalability), and (3) the scope of optimization variables considered (multi-objective capability). Furthermore, existing methods often fall short of comprehensively incorporating the factors related to the mRNA lifecycle and translation process, including intrinsic mRNA sequence properties, secondary structure, translation elongation kinetics, and tRNA availability. To address these limitations, we introduce \textbf{RNop}, a novel deep learning-based method for mRNA optimization. We collect a large-scale dataset containing over 3 million sequences and design four specialized loss functions, the GPLoss, CAILoss, tAILoss, and MFELoss, which simultaneously enable explicit control over sequence fidelity while optimizing species-specific codon adaptation, tRNA availability, and desirable mRNA secondary structure features. Then, we demonstrate RNop's effectiveness through extensive in silico and in vivo experiments. RNop ensures high sequence fidelity, achieves significant computational throughput up to 47.32 sequences/s, and yields optimized mRNA sequences resulting in a significant increase in protein expression for functional proteins compared to controls. RNop surpasses current methodologies in both quantitative metrics and experimental validation, enlightening a new dawn for efficient and effective mRNA design. Code and models will be available at https://github.com/HudenJear/RPLoss.
AI-driven Prediction of Insulin Resistance in Normal Populations: Comparing Models and Criteria
Mar 07, 2025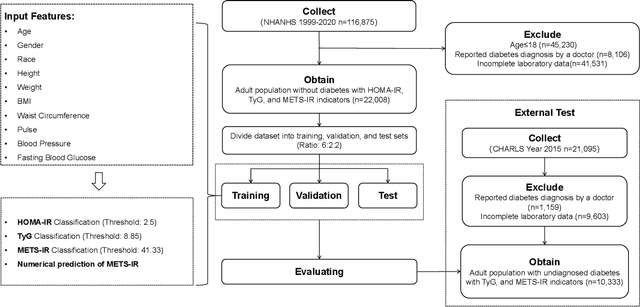
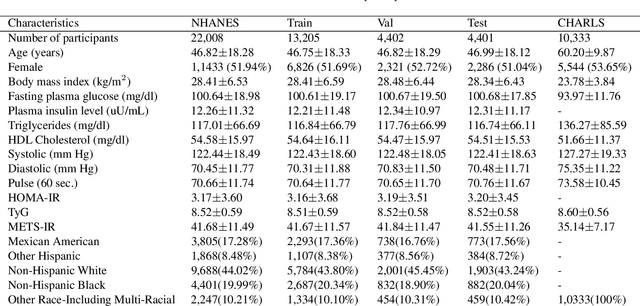
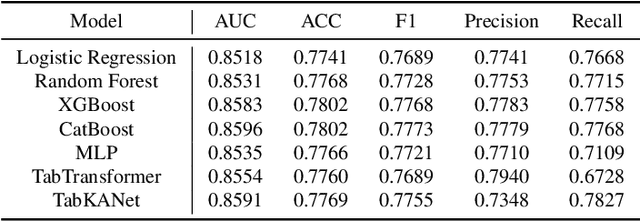
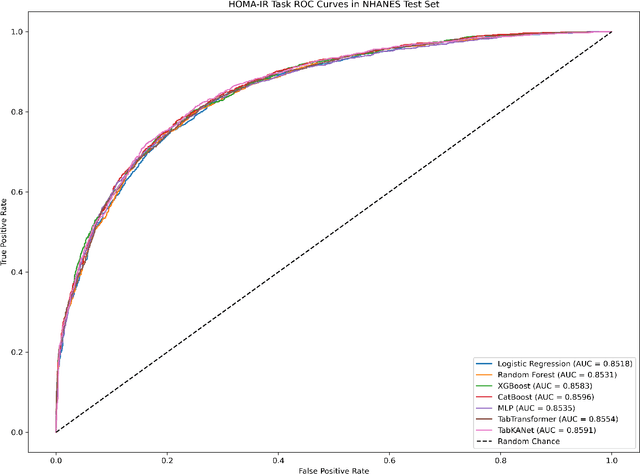
Insulin resistance (IR) is a key precursor to diabetes and a significant risk factor for cardiovascular disease. Traditional IR assessment methods require multiple blood tests. We developed a simple AI model using only fasting blood glucose to predict IR in non-diabetic populations. Data from the NHANES (1999-2020) and CHARLS (2015) studies were used for model training and validation. Input features included age, gender, height, weight, blood pressure, waist circumference, and fasting blood glucose. The CatBoost algorithm achieved AUC values of 0.8596 (HOMA-IR) and 0.7777 (TyG index) in NHANES, with an external AUC of 0.7442 for TyG. For METS-IR prediction, the model achieved AUC values of 0.9731 (internal) and 0.9591 (external), with RMSE values of 3.2643 (internal) and 3.057 (external). SHAP analysis highlighted waist circumference as a key predictor of IR. This AI model offers a minimally invasive and effective tool for IR prediction, supporting early diabetes and cardiovascular disease prevention.
Acquire Precise and Comparable Fundus Image Quality Score: FTHNet and FQS Dataset
Nov 19, 2024The retinal fundus images are utilized extensively in the diagnosis, and their quality can directly affect the diagnosis results. However, due to the insufficient dataset and algorithm application, current fundus image quality assessment (FIQA) methods are not powerful enough to meet ophthalmologists` demands. In this paper, we address the limitations of datasets and algorithms in FIQA. First, we establish a new FIQA dataset, Fundus Quality Score(FQS), which includes 2246 fundus images with two labels: a continuous Mean Opinion Score varying from 0 to 100 and a three-level quality label. Then, we propose a FIQA Transformer-based Hypernetwork (FTHNet) to solve these tasks with regression results rather than classification results in conventional FIQA works. The FTHNet is optimized for the FIQA tasks with extensive experiments. Results on our FQS dataset show that the FTHNet can give quality scores for fundus images with PLCC of 0.9423 and SRCC of 0.9488, significantly outperforming other methods with fewer parameters and less computation complexity.We successfully build a dataset and model addressing the problems of current FIQA methods. Furthermore, the model deployment experiments demonstrate its potential in automatic medical image quality control. All experiments are carried out with 10-fold cross-validation to ensure the significance of the results.
Versatile Cataract Fundus Image Restoration Model Utilizing Unpaired Cataract and High-quality Images
Nov 19, 2024



Cataract is one of the most common blinding eye diseases and can be treated by surgery. However, because cataract patients may also suffer from other blinding eye diseases, ophthalmologists must diagnose them before surgery. The cloudy lens of cataract patients forms a hazy degeneration in the fundus images, making it challenging to observe the patient's fundus vessels, which brings difficulties to the diagnosis process. To address this issue, this paper establishes a new cataract image restoration method named Catintell. It contains a cataract image synthesizing model, Catintell-Syn, and a restoration model, Catintell-Res. Catintell-Syn uses GAN architecture with fully unsupervised data to generate paired cataract-like images with realistic style and texture rather than the conventional Gaussian degradation algorithm. Meanwhile, Catintell-Res is an image restoration network that can improve the quality of real cataract fundus images using the knowledge learned from synthetic cataract images. Extensive experiments show that Catintell-Res outperforms other cataract image restoration methods in PSNR with 39.03 and SSIM with 0.9476. Furthermore, the universal restoration ability that Catintell-Res gained from unpaired cataract images can process cataract images from various datasets. We hope the models can help ophthalmologists identify other blinding eye diseases of cataract patients and inspire more medical image restoration methods in the future.
TabKANet: Tabular Data Modelling with Kolmogorov-Arnold Network and Transformer
Sep 13, 2024



Tabular data is the most common type of data in real-life scenarios. In this study, we propose a method based on the TabKANet architecture, which utilizes the Kolmogorov-Arnold network to encode numerical features and merge them with categorical features, enabling unified modeling of tabular data on the Transformer architecture. This model demonstrates outstanding performance in six widely used binary classification tasks, suggesting that TabKANet has the potential to become a standard approach for tabular modeling, surpassing traditional neural networks. Furthermore, this research reveals the significant advantages of the Kolmogorov-Arnold network in encoding numerical features. The code of our work is available at https://github.com/tsinghuamedgao20/TabKANet.