 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSupervised Pretraining for Molecular Force Fields and Properties Prediction
Paper and Code
Nov 23, 2022
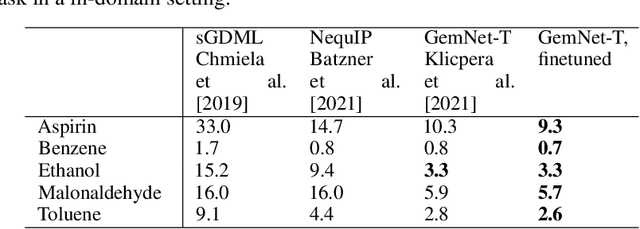
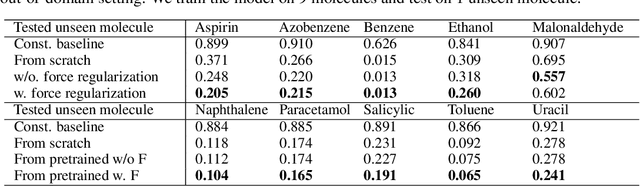
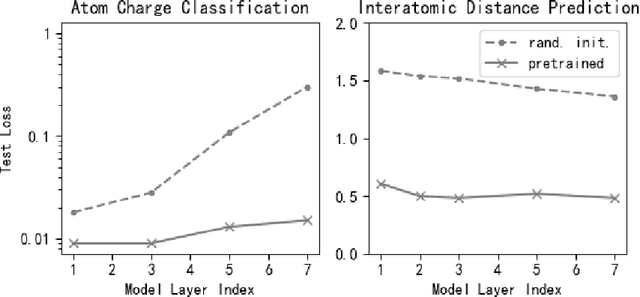
Machine learning approaches have become popular for molecular modeling tasks, including molecular force fields and properties prediction. Traditional supervised learning methods suffer from scarcity of labeled data for particular tasks, motivating the use of large-scale dataset for other relevant tasks. We propose to pretrain neural networks on a dataset of 86 millions of molecules with atom charges and 3D geometries as inputs and molecular energies as labels. Experiments show that, compared to training from scratch, fine-tuning the pretrained model can significantly improve the performance for seven molecular property prediction tasks and two force field tasks. We also demonstrate that the learned representations from the pretrained model contain adequate information about molecular structures, by showing that linear probing of the representations can predict many molecular information including atom types, interatomic distances, class of molecular scaffolds, and existence of molecular fragments. Our results show that supervised pretraining is a promising research direction in molecular modeling