 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLate-to-Early Training: LET LLMs Learn Earlier, So Faster and Better
Feb 05, 2026As Large Language Models (LLMs) achieve remarkable empirical success through scaling model and data size, pretraining has become increasingly critical yet computationally prohibitive, hindering rapid development. Despite the availability of numerous pretrained LLMs developed at significant computational expense, a fundamental real-world question remains underexplored: \textit{Can we leverage existing small pretrained models to accelerate the training of larger models?} In this paper, we propose a Late-to-Early Training (LET) paradigm that enables LLMs to explicitly learn later knowledge in earlier steps and earlier layers. The core idea is to guide the early layers of an LLM during early training using representations from the late layers of a pretrained (i.e. late training phase) model. We identify two key mechanisms that drive LET's effectiveness: late-to-early-step learning and late-to-early-layer learning. These mechanisms significantly accelerate training convergence while robustly enhancing both language modeling capabilities and downstream task performance, enabling faster training with superior performance. Extensive experiments on 1.4B and 7B parameter models demonstrate LET's efficiency and effectiveness. Notably, when training a 1.4B LLM on the Pile dataset, our method achieves up to 1.6$\times$ speedup with nearly 5\% improvement in downstream task accuracy compared to standard training, even when using a pretrained model with 10$\times$ fewer parameters than the target model.
SPARKLING: Balancing Signal Preservation and Symmetry Breaking for Width-Progressive Learning
Feb 02, 2026Progressive Learning (PL) reduces pre-training computational overhead by gradually increasing model scale. While prior work has extensively explored depth expansion, width expansion remains significantly understudied, with the few existing methods limited to the early stages of training. However, expanding width during the mid-stage is essential for maximizing computational savings, yet it remains a formidable challenge due to severe training instabilities. Empirically, we show that naive initialization at this stage disrupts activation statistics, triggering loss spikes, while copy-based initialization introduces gradient symmetry that hinders feature diversity. To address these issues, we propose SPARKLING (balancing {S}ignal {P}reservation {A}nd symmet{R}y brea{K}ing for width-progressive {L}earn{ING}), a novel framework for mid-stage width expansion. Our method achieves signal preservation via RMS-scale consistency, stabilizing activation statistics during expansion. Symmetry breaking is ensured through asymmetric optimizer state resetting and learning rate re-warmup. Extensive experiments on Mixture-of-Experts (MoE) models demonstrate that, across multiple width axes and optimizer families, SPARKLING consistently outperforms training from scratch and reduces training cost by up to 35% under $2\times$ width expansion.
Seedance 1.5 pro: A Native Audio-Visual Joint Generation Foundation Model
Dec 23, 2025Recent strides in video generation have paved the way for unified audio-visual generation. In this work, we present Seedance 1.5 pro, a foundational model engineered specifically for native, joint audio-video generation. Leveraging a dual-branch Diffusion Transformer architecture, the model integrates a cross-modal joint module with a specialized multi-stage data pipeline, achieving exceptional audio-visual synchronization and superior generation quality. To ensure practical utility, we implement meticulous post-training optimizations, including Supervised Fine-Tuning (SFT) on high-quality datasets and Reinforcement Learning from Human Feedback (RLHF) with multi-dimensional reward models. Furthermore, we introduce an acceleration framework that boosts inference speed by over 10X. Seedance 1.5 pro distinguishes itself through precise multilingual and dialect lip-syncing, dynamic cinematic camera control, and enhanced narrative coherence, positioning it as a robust engine for professional-grade content creation. Seedance 1.5 pro is now accessible on Volcano Engine at https://console.volcengine.com/ark/region:ark+cn-beijing/experience/vision?type=GenVideo.
Virtual Width Networks
Nov 17, 2025



We introduce Virtual Width Networks (VWN), a framework that delivers the benefits of wider representations without incurring the quadratic cost of increasing the hidden size. VWN decouples representational width from backbone width, expanding the embedding space while keeping backbone compute nearly constant. In our large-scale experiment, an 8-times expansion accelerates optimization by over 2 times for next-token and 3 times for next-2-token prediction. The advantage amplifies over training as both the loss gap grows and the convergence-speedup ratio increases, showing that VWN is not only token-efficient but also increasingly effective with scale. Moreover, we identify an approximately log-linear scaling relation between virtual width and loss reduction, offering an initial empirical basis and motivation for exploring virtual-width scaling as a new dimension of large-model efficiency.
Model Merging in Pre-training of Large Language Models
May 17, 2025Model merging has emerged as a promising technique for enhancing large language models, though its application in large-scale pre-training remains relatively unexplored. In this paper, we present a comprehensive investigation of model merging techniques during the pre-training process. Through extensive experiments with both dense and Mixture-of-Experts (MoE) architectures ranging from millions to over 100 billion parameters, we demonstrate that merging checkpoints trained with constant learning rates not only achieves significant performance improvements but also enables accurate prediction of annealing behavior. These improvements lead to both more efficient model development and significantly lower training costs. Our detailed ablation studies on merging strategies and hyperparameters provide new insights into the underlying mechanisms while uncovering novel applications. Through comprehensive experimental analysis, we offer the open-source community practical pre-training guidelines for effective model merging.
Seed1.5-VL Technical Report
May 11, 2025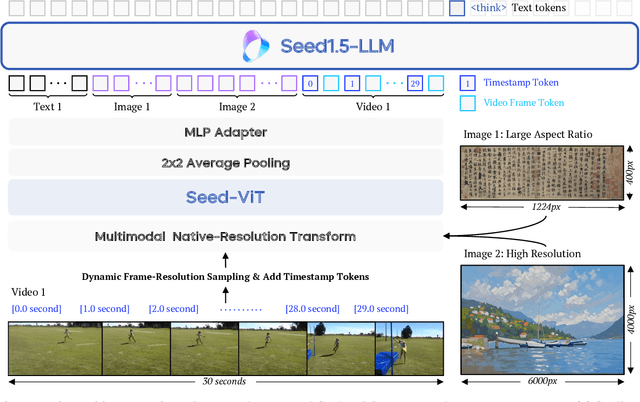


We present Seed1.5-VL, a vision-language foundation model designed to advance general-purpose multimodal understanding and reasoning. Seed1.5-VL is composed with a 532M-parameter vision encoder and a Mixture-of-Experts (MoE) LLM of 20B active parameters. Despite its relatively compact architecture, it delivers strong performance across a wide spectrum of public VLM benchmarks and internal evaluation suites, achieving the state-of-the-art performance on 38 out of 60 public benchmarks. Moreover, in agent-centric tasks such as GUI control and gameplay, Seed1.5-VL outperforms leading multimodal systems, including OpenAI CUA and Claude 3.7. Beyond visual and video understanding, it also demonstrates strong reasoning abilities, making it particularly effective for multimodal reasoning challenges such as visual puzzles. We believe these capabilities will empower broader applications across diverse tasks. In this report, we mainly provide a comprehensive review of our experiences in building Seed1.5-VL across model design, data construction, and training at various stages, hoping that this report can inspire further research. Seed1.5-VL is now accessible at https://www.volcengine.com/ (Volcano Engine Model ID: doubao-1-5-thinking-vision-pro-250428)
Multi-SWE-bench: A Multilingual Benchmark for Issue Resolving
Apr 03, 2025The task of issue resolving is to modify a codebase to generate a patch that addresses a given issue. However, existing benchmarks, such as SWE-bench, focus almost exclusively on Python, making them insufficient for evaluating Large Language Models (LLMs) across diverse software ecosystems. To address this, we introduce a multilingual issue-resolving benchmark, called Multi-SWE-bench, covering Java, TypeScript, JavaScript, Go, Rust, C, and C++. It includes a total of 1,632 high-quality instances, which were carefully annotated from 2,456 candidates by 68 expert annotators, ensuring that the benchmark can provide an accurate and reliable evaluation. Based on Multi-SWE-bench, we evaluate a series of state-of-the-art models using three representative methods (Agentless, SWE-agent, and OpenHands) and present a comprehensive analysis with key empirical insights. In addition, we launch a Multi-SWE-RL open-source community, aimed at building large-scale reinforcement learning (RL) training datasets for issue-resolving tasks. As an initial contribution, we release a set of 4,723 well-structured instances spanning seven programming languages, laying a solid foundation for RL research in this domain. More importantly, we open-source our entire data production pipeline, along with detailed tutorials, encouraging the open-source community to continuously contribute and expand the dataset. We envision our Multi-SWE-bench and the ever-growing Multi-SWE-RL community as catalysts for advancing RL toward its full potential, bringing us one step closer to the dawn of AGI.
FullStack Bench: Evaluating LLMs as Full Stack Coders
Dec 03, 2024



As the capabilities of code large language models (LLMs) continue to expand, their applications across diverse code intelligence domains are rapidly increasing. However, most existing datasets only evaluate limited application domains. To address this gap, we have developed a comprehensive code evaluation dataset FullStack Bench focusing on full-stack programming, which encompasses a wide range of application domains (e.g., basic programming, data analysis, software engineering, mathematics, and machine learning). Besides, to assess multilingual programming capabilities, in FullStack Bench, we design real-world instructions and corresponding unit test cases from 16 widely-used programming languages to reflect real-world usage scenarios rather than simple translations. Moreover, we also release an effective code sandbox execution tool (i.e., SandboxFusion) supporting various programming languages and packages to evaluate the performance of our FullStack Bench efficiently. Comprehensive experimental results on our FullStack Bench demonstrate the necessity and effectiveness of our FullStack Bench and SandboxFusion.
Unlock the Correlation between Supervised Fine-Tuning and Reinforcement Learning in Training Code Large Language Models
Jun 14, 2024



Automatic code generation has been a longstanding research topic. With the advancement of general-purpose large language models (LLMs), the ability to code stands out as one important measure to the model's reasoning performance. Usually, a two-stage training paradigm is implemented to obtain a Code LLM, namely the pretraining and the fine-tuning. Within the fine-tuning, supervised fine-tuning (SFT), and reinforcement learning (RL) are often used to improve the model's zero-shot ability. A large number of work has been conducted to improve the model's performance on code-related benchmarks with either modifications to the algorithm or refinement of the dataset. However, we still lack a deep insight into the correlation between SFT and RL. For instance, what kind of dataset should be used to ensure generalization, or what if we abandon the SFT phase in fine-tuning. In this work, we make an attempt to understand the correlation between SFT and RL. To facilitate our research, we manually craft 100 basis python functions, called atomic functions, and then a synthesizing pipeline is deployed to create a large number of synthetic functions on top of the atomic ones. In this manner, we ensure that the train and test sets remain distinct, preventing data contamination. Through comprehensive ablation study, we find: (1) Both atomic and synthetic functions are indispensable for SFT's generalization, and only a handful of synthetic functions are adequate; (2) Through RL, the SFT's generalization to target domain can be greatly enhanced, even with the same training prompts; (3) Training RL from scratch can alleviate the over-fitting issue introduced in the SFT phase.
BAMBOO: a predictive and transferable machine learning force field framework for liquid electrolyte development
Apr 12, 2024Despite the widespread applications of machine learning force field (MLFF) on solids and small molecules, there is a notable gap in applying MLFF to complex liquid electrolytes. In this work, we introduce BAMBOO (ByteDance AI Molecular Simulation Booster), a novel framework for molecular dynamics (MD) simulations, with a demonstration of its capabilities in the context of liquid electrolytes for lithium batteries. We design a physics-inspired graph equivariant transformer architecture as the backbone of BAMBOO to learn from quantum mechanical simulations. Additionally, we pioneer an ensemble knowledge distillation approach and apply it on MLFFs to improve the stability of MD simulations. Finally, we propose the density alignment algorithm to align BAMBOO with experimental measurements. BAMBOO demonstrates state-of-the-art accuracy in predicting key electrolyte properties such as density, viscosity, and ionic conductivity across various solvents and salt combinations. Our current model, trained on more than 15 chemical species, achieves the average density error of 0.01 g/cm$^3$ on various compositions compared with experimental data. Moreover, our model demonstrates transferability to molecules not included in the quantum mechanical dataset. We envision this work as paving the way to a "universal MLFF" capable of simulating properties of common organic liquids.