 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtenix-Mini: Efficient Structure Predictor via Compact Architecture, Few-Step Diffusion and Switchable pLM
Jul 16, 2025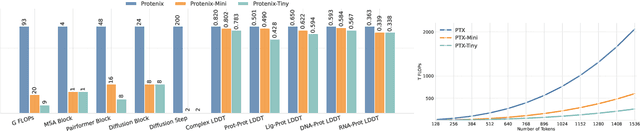
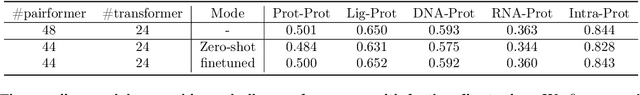
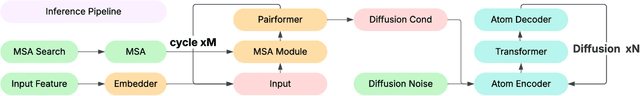
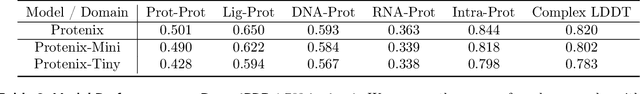
Lightweight inference is critical for biomolecular structure prediction and other downstream tasks, enabling efficient real-world deployment and inference-time scaling for large-scale applications. In this work, we address the challenge of balancing model efficiency and prediction accuracy by making several key modifications, 1) Multi-step AF3 sampler is replaced by a few-step ODE sampler, significantly reducing computational overhead for the diffusion module part during inference; 2) In the open-source Protenix framework, a subset of pairformer or diffusion transformer blocks doesn't make contributions to the final structure prediction, presenting opportunities for architectural pruning and lightweight redesign; 3) A model incorporating an ESM module is trained to substitute the conventional MSA module, reducing MSA preprocessing time. Building on these key insights, we present Protenix-Mini, a compact and optimized model designed for efficient protein structure prediction. This streamlined version incorporates a more efficient architectural design with a two-step Ordinary Differential Equation (ODE) sampling strategy. By eliminating redundant Transformer components and refining the sampling process, Protenix-Mini significantly reduces model complexity with slight accuracy drop. Evaluations on benchmark datasets demonstrate that it achieves high-fidelity predictions, with only a negligible 1 to 5 percent decrease in performance on benchmark datasets compared to its full-scale counterpart. This makes Protenix-Mini an ideal choice for applications where computational resources are limited but accurate structure prediction remains crucial.
Learning Regularized Positional Encoding for Molecular Prediction
Nov 23, 2022
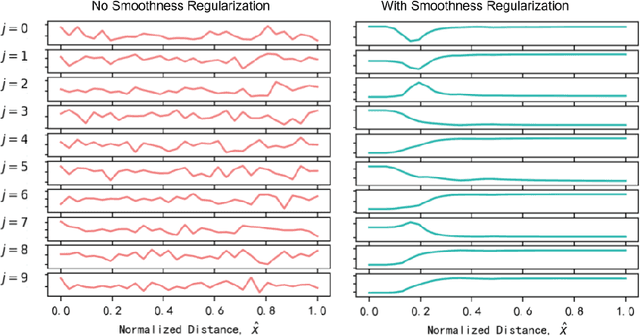


Machine learning has become a promising approach for molecular modeling. Positional quantities, such as interatomic distances and bond angles, play a crucial role in molecule physics. The existing works rely on careful manual design of their representation. To model the complex nonlinearity in predicting molecular properties in an more end-to-end approach, we propose to encode the positional quantities with a learnable embedding that is continuous and differentiable. A regularization technique is employed to encourage embedding smoothness along the physical dimension. We experiment with a variety of molecular property and force field prediction tasks. Improved performance is observed for three different model architectures after plugging in the proposed positional encoding method. In addition, the learned positional encoding allows easier physics-based interpretation. We observe that tasks of similar physics have the similar learned positional encoding.
Supervised Pretraining for Molecular Force Fields and Properties Prediction
Nov 23, 2022
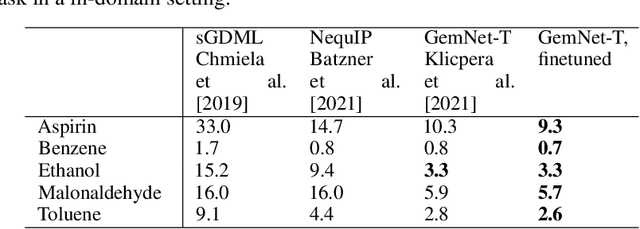
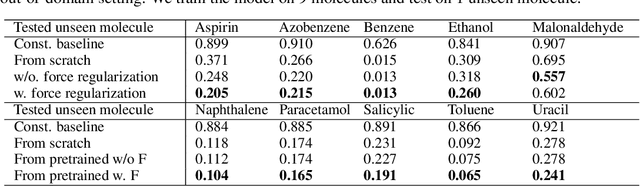
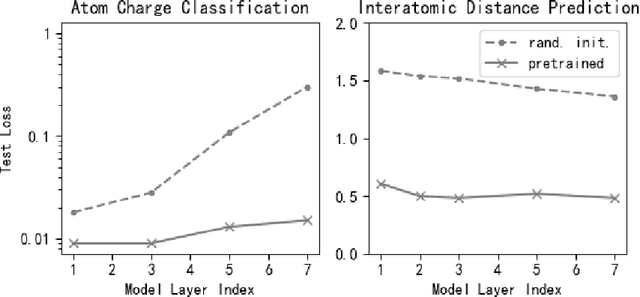
Machine learning approaches have become popular for molecular modeling tasks, including molecular force fields and properties prediction. Traditional supervised learning methods suffer from scarcity of labeled data for particular tasks, motivating the use of large-scale dataset for other relevant tasks. We propose to pretrain neural networks on a dataset of 86 millions of molecules with atom charges and 3D geometries as inputs and molecular energies as labels. Experiments show that, compared to training from scratch, fine-tuning the pretrained model can significantly improve the performance for seven molecular property prediction tasks and two force field tasks. We also demonstrate that the learned representations from the pretrained model contain adequate information about molecular structures, by showing that linear probing of the representations can predict many molecular information including atom types, interatomic distances, class of molecular scaffolds, and existence of molecular fragments. Our results show that supervised pretraining is a promising research direction in molecular modeling
Deep Retrieval: An End-to-End Learnable Structure Model for Large-Scale Recommendations
Jul 12, 2020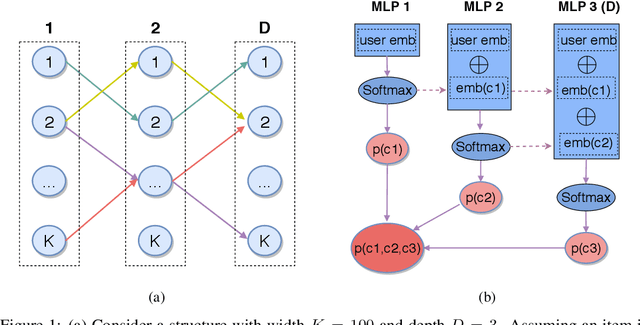
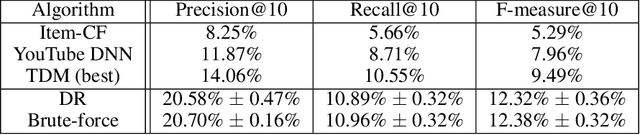
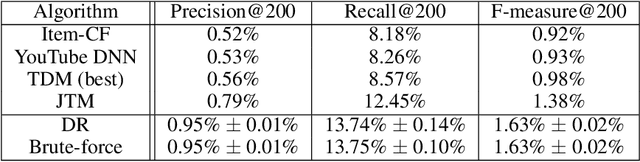
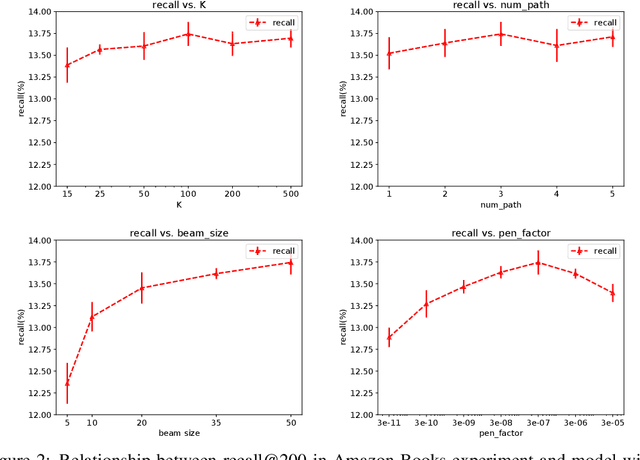
One of the core problems in large-scale recommendations is to retrieve top relevant candidates accurately and efficiently, preferably in sub-linear time. Previous approaches are mostly based on a two-step procedure: first learn an inner-product model and then use maximum inner product search (MIPS) algorithms to search top candidates, leading to potential loss of retrieval accuracy. In this paper, we present Deep Retrieval (DR), an end-to-end learnable structure model for large-scale recommendations. DR encodes all candidates into a discrete latent space. Those latent codes for the candidates are model parameters and to be learnt together with other neural network parameters to maximize the same objective function. With the model learnt, a beam search over the latent codes is performed to retrieve the top candidates. Empirically, we showed that DR, with sub-linear computational complexity, can achieve almost the same accuracy as the brute-force baseline.