 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeViral Proteins Reveal Geometry of Protein Language Models
Jun 10, 2026Protein language models are trained on highly imbalanced datasets, raising the question of how they represent underrepresented biological sequences. Using viral proteins as a case study across ESM model families, we identify a dominant nativeness axis in embedding space, aligned with masked reconstruction perplexity, that orders sequences from well-modeled cellular proteins through viral proteins to shuffled and random sequences. Scaling contracts this axis unevenly across viral families. Despite this, protein language model embeddings retain viral-specific signal: viral proteins remain linearly separable beyond zero-shot perplexity and shallow sequence features. Together, these results suggest that pLM representations are structured by a general notion of nativeness while preserving information specific to distinct biological groups.
Computational Design and Experimental Validation of Photoactive PARP1 Inhibitors
Apr 27, 2026Light-activated drugs are a promising way to treat localized diseases for which existing treatments have severe side effects. However, their development is complicated by the set of photophysical and biological properties that must be simultaneously optimized. Here we used computational techniques to find a set of promising candidates for the photoactive inhibition of the poly(ADP-ribose) polymerase 1 (PARP1) cancer target. Using our recently developed methods based on atomistic simulation and machine learning (ML), we screened a set of 5 million hypothetical photoactive ligands. Our workflow used protein-ligand docking to identify candidates with differential PARP1 binding under light and dark conditions; ML force fields and quantum chemistry calculations to predict p$K_\mathrm{a}$, absorption spectra, and thermal half-lives; graph-based surrogate models to screen additional compounds; excited-state nonadiabatic dynamics with ML force fields to estimate quantum yields; and free energy perturbation (FEP) to refine binding predictions. From these predictions, we prioritized a small set of synthetically feasible candidates expected to have red-shifted absorption spectra, thermal half-lives on the order of seconds to minutes, and isomer-dependent PARP1 binding under visible-light control. We synthesized 10 candidates and experimentally characterized their photobehavior and PARP1 inhibition constants. Among the validated compounds, \textbf{1} showed a 15-fold increase in inhibition of PARP1 upon green-light irradiation at 519 nm (208.8 $\pm$ 28.3 $μ$M vs 14.4 $\pm$ 1.9 $μ$M). These results validate the computation-guided screening strategy for identifying red-shifted PARP1 photoinhibitors, while also underscoring current limitations such as rapid thermal relaxation in aqueous media.
Thermal half-lives of azobenzene derivatives: virtual screening based on intersystem crossing using a machine learning potential
Jul 26, 2022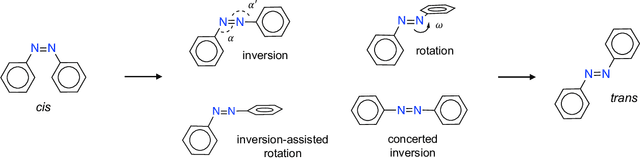
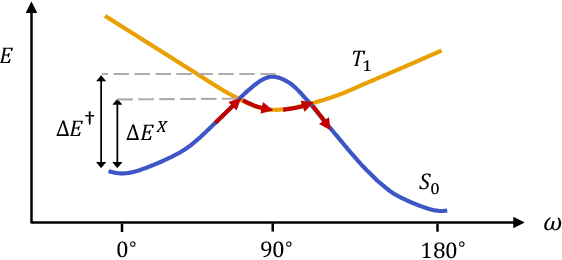
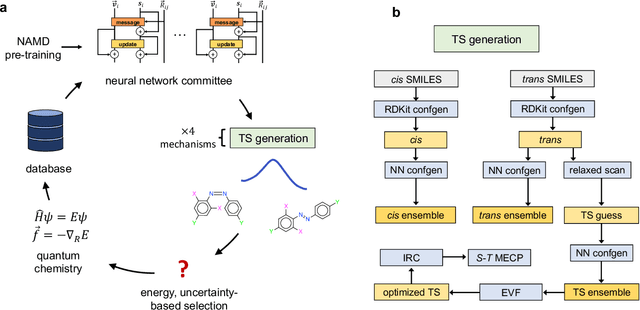
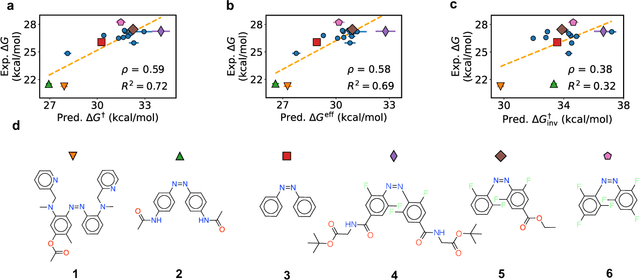
Molecular photoswitches are the foundation of light-activated drugs. A key photoswitch is azobenzene, which exhibits trans-cis isomerism in response to light. The thermal half-life of the cis isomer is of crucial importance, since it controls the duration of the light-induced biological effect. Here we introduce a computational tool for predicting the thermal half-lives of azobenzene derivatives. Our automated approach uses a fast and accurate machine learning potential trained on quantum chemistry data. Building on well-established earlier evidence, we argue that thermal isomerization proceeds through rotation mediated by intersystem crossing, and incorporate this mechanism into our automated workflow. We use our approach to predict the thermal half-lives of 19,000 azobenzene derivatives. We explore trends and tradeoffs between barriers and absorption wavelengths, and open-source our data and software to accelerate research in photopharmacology.
Excited state, non-adiabatic dynamics of large photoswitchable molecules using a chemically transferable machine learning potential
Aug 10, 2021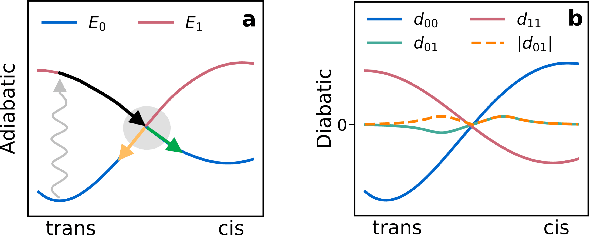
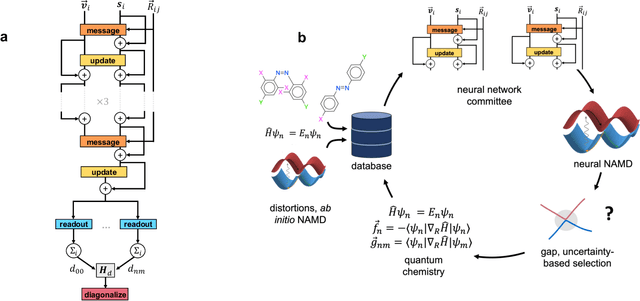
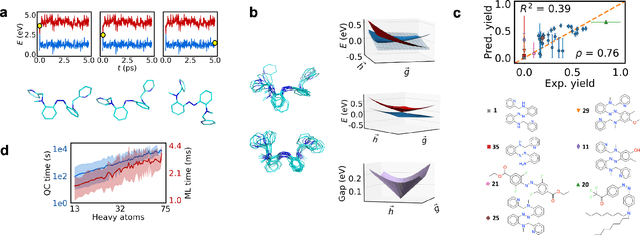
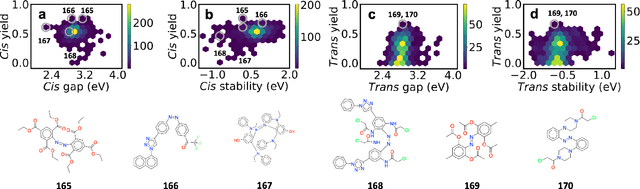
Light-induced chemical processes are ubiquitous in nature and have widespread technological applications. For example, the photoisomerization of azobenzene allows a drug with an azo scaffold to be activated with light. In principle, photoswitches with useful reactive properties, such as high isomerization yields, can be identified through virtual screening with reactive simulations. In practice these simulations are rarely used for screening, since they require hundreds of trajectories and expensive quantum chemical methods to account for non-adiabatic excited state effects. Here we introduce a neural network potential to accelerate such simulations for azobenzene derivatives. The model, which is based on diabatic states, is called the \textit{diabatic artificial neural network} (DANN). The network is six orders of magnitude faster than the quantum chemistry method used for training. DANN is transferable to molecules outside the training set, predicting quantum yields for unseen species that are correlated with experiment. We use the model to virtually screen 3,100 hypothetical molecules, and identify several species with extremely high quantum yields. Our results pave the way for fast and accurate virtual screening of photoactive compounds.