 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGenerative structural elucidation from mass spectra as an iterative optimization problem
Feb 07, 2026Liquid chromatography tandem mass spectrometry (LC-MS/MS) is a critical analytical technique for molecular identification across metabolomics, environmental chemistry, and chemical forensics. A variety of computational methods have emerged for structural annotation of spectral features of interest, but many of these features cannot be confidently annotated with reference structures or spectra. Here, we introduce FOAM (Formula-constrained Optimization for Annotating Metabolites), a computational workflow that poses structure elucidation from LC-MS/MS as an iterative optimization problem. FOAM couples a formula-constrained graph genetic algorithm with spectral simulation to explore candidate annotations given an experimental spectrum. We demonstrate FOAM's performance on the NIST'20 and MassSpecGym datasets as both a standalone elucidation pipeline and as a complement to existing inverse models. This work establishes iterative optimization as an effective and extensible paradigm for structural elucidation.
Single-model uncertainty quantification in neural network potentials does not consistently outperform model ensembles
May 02, 2023Neural networks (NNs) often assign high confidence to their predictions, even for points far out-of-distribution, making uncertainty quantification (UQ) a challenge. When they are employed to model interatomic potentials in materials systems, this problem leads to unphysical structures that disrupt simulations, or to biased statistics and dynamics that do not reflect the true physics. Differentiable UQ techniques can find new informative data and drive active learning loops for robust potentials. However, a variety of UQ techniques, including newly developed ones, exist for atomistic simulations and there are no clear guidelines for which are most effective or suitable for a given case. In this work, we examine multiple UQ schemes for improving the robustness of NN interatomic potentials (NNIPs) through active learning. In particular, we compare incumbent ensemble-based methods against strategies that use single, deterministic NNs: mean-variance estimation, deep evidential regression, and Gaussian mixture models. We explore three datasets ranging from in-domain interpolative learning to more extrapolative out-of-domain generalization challenges: rMD17, ammonia inversion, and bulk silica glass. Performance is measured across multiple metrics relating model error to uncertainty. Our experiments show that none of the methods consistently outperformed each other across the various metrics. Ensembling remained better at generalization and for NNIP robustness; MVE only proved effective for in-domain interpolation, while GMM was better out-of-domain; and evidential regression, despite its promise, was not the preferable alternative in any of the cases. More broadly, cost-effective, single deterministic models cannot yet consistently match or outperform ensembling for uncertainty quantification in NNIPs.
Prefix-tree Decoding for Predicting Mass Spectra from Molecules
Mar 11, 2023
Computational predictions of mass spectra from molecules have enabled the discovery of clinically relevant metabolites. However, such predictive tools are still limited as they occupy one of two extremes, either operating (a) by fragmenting molecules combinatorially with overly rigid constraints on potential rearrangements and poor time complexity or (b) by decoding lossy and nonphysical discretized spectra vectors. In this work, we introduce a new intermediate strategy for predicting mass spectra from molecules by treating mass spectra as sets of chemical formulae, which are themselves multisets of atoms. After first encoding an input molecular graph, we decode a set of chemical subformulae, each of which specify a predicted peak in the mass spectra, the intensities of which are predicted by a second model. Our key insight is to overcome the combinatorial possibilities for chemical subformulae by decoding the formula set using a prefix tree structure, atom-type by atom-type, representing a general method for ordered multiset decoding. We show promising empirical results on mass spectra prediction tasks.
Machine learning modeling of family wide enzyme-substrate specificity screens
Sep 08, 2021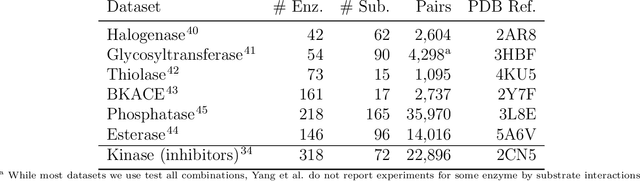
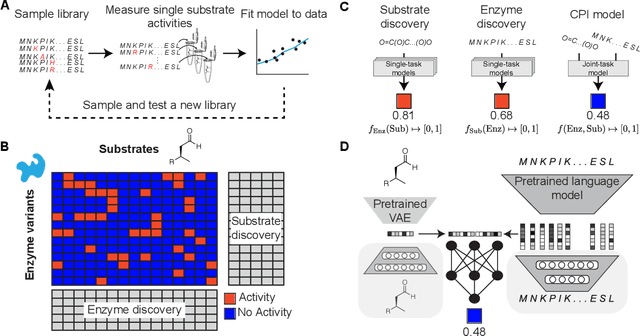
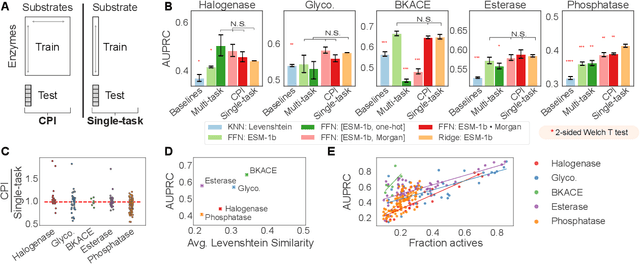
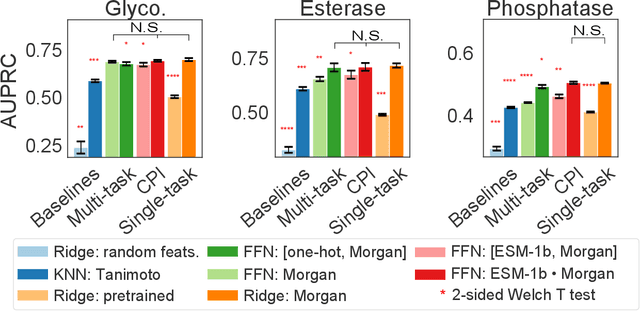
Biocatalysis is a promising approach to sustainably synthesize pharmaceuticals, complex natural products, and commodity chemicals at scale. However, the adoption of biocatalysis is limited by our ability to select enzymes that will catalyze their natural chemical transformation on non-natural substrates. While machine learning and in silico directed evolution are well-posed for this predictive modeling challenge, efforts to date have primarily aimed to increase activity against a single known substrate, rather than to identify enzymes capable of acting on new substrates of interest. To address this need, we curate 6 different high-quality enzyme family screens from the literature that each measure multiple enzymes against multiple substrates. We compare machine learning-based compound-protein interaction (CPI) modeling approaches from the literature used for predicting drug-target interactions. Surprisingly, comparing these interaction-based models against collections of independent (single task) enzyme-only or substrate-only models reveals that current CPI approaches are incapable of learning interactions between compounds and proteins in the current family level data regime. We further validate this observation by demonstrating that our no-interaction baseline can outperform CPI-based models from the literature used to guide the discovery of kinase inhibitors. Given the high performance of non-interaction based models, we introduce a new structure-based strategy for pooling residue representations across a protein sequence. Altogether, this work motivates a principled path forward in order to build and evaluate meaningful predictive models for biocatalysis and other drug discovery applications.
Improved Conditional Flow Models for Molecule to Image Synthesis
Jun 15, 2020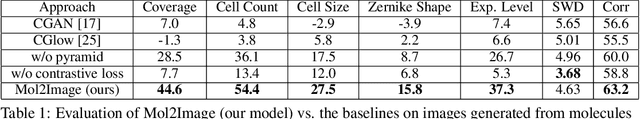
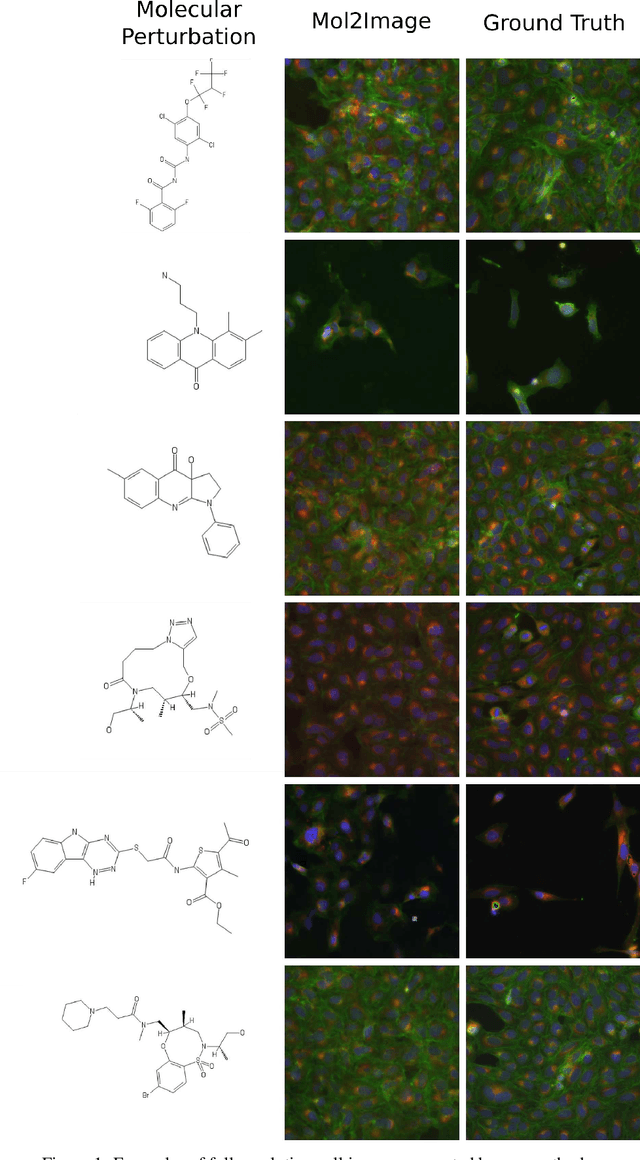
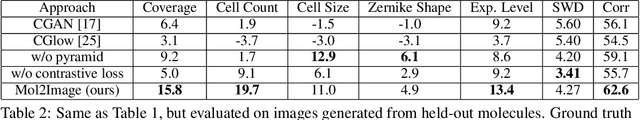
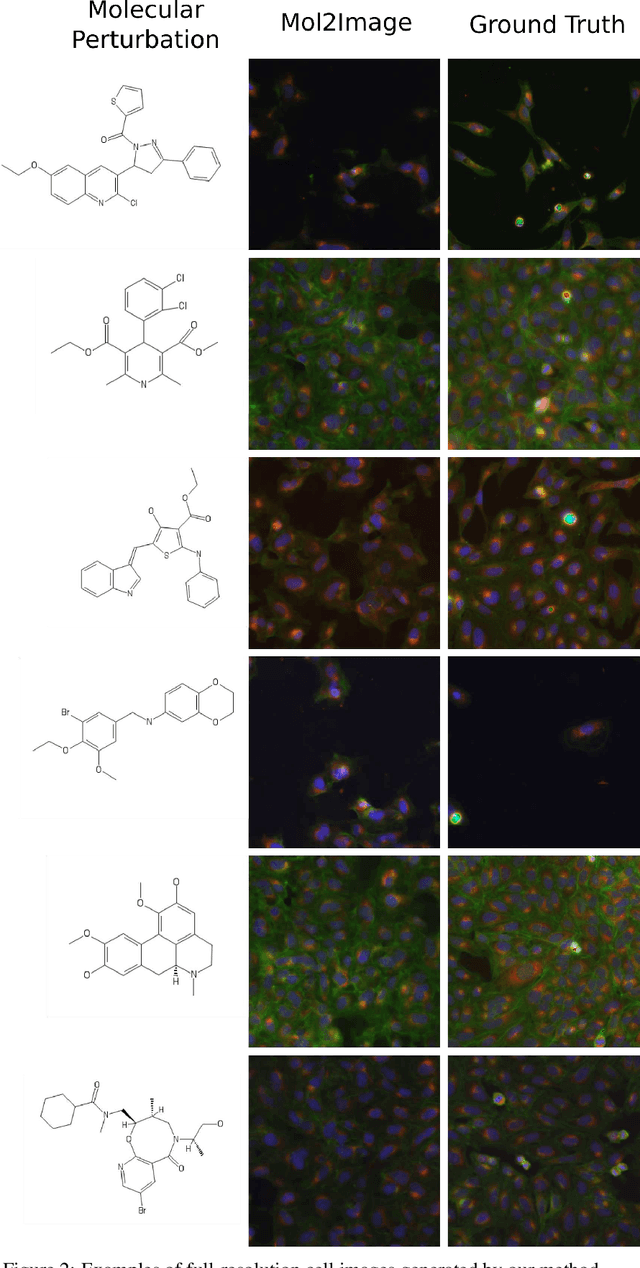
In this paper, we aim to synthesize cell microscopy images under different molecular interventions, motivated by practical applications to drug development. Building on the recent success of graph neural networks for learning molecular embeddings and flow-based models for image generation, we propose Mol2Image: a flow-based generative model for molecule to cell image synthesis. To generate cell features at different resolutions and scale to high-resolution images, we develop a novel multi-scale flow architecture based on a Haar wavelet image pyramid. To maximize the mutual information between the generated images and the molecular interventions, we devise a training strategy based on contrastive learning. To evaluate our model, we propose a new set of metrics for biological image generation that are robust, interpretable, and relevant to practitioners. We show quantitatively that our method learns a meaningful embedding of the molecular intervention, which is translated into an image representation reflecting the biological effects of the intervention.