 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePrefix-tree Decoding for Predicting Mass Spectra from Molecules
Paper and Code
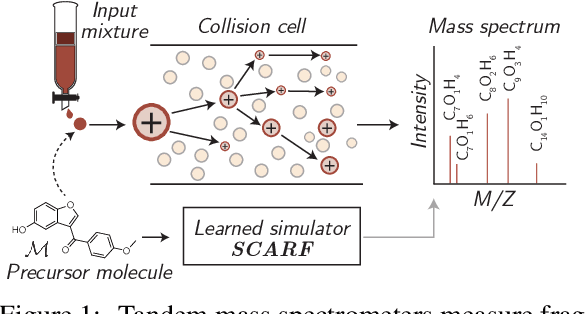
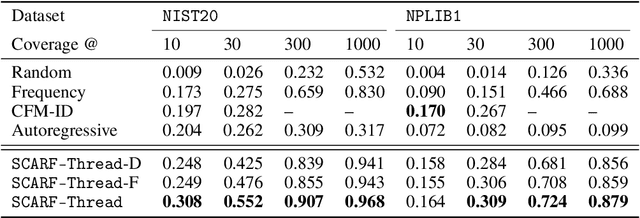
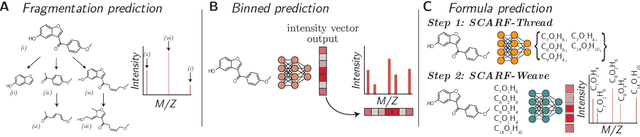
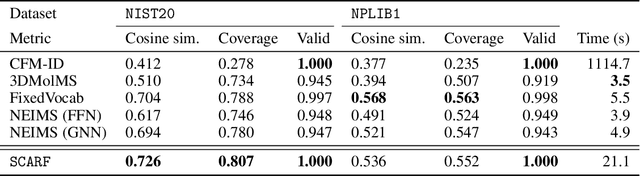
Computational predictions of mass spectra from molecules have enabled the discovery of clinically relevant metabolites. However, such predictive tools are still limited as they occupy one of two extremes, either operating (a) by fragmenting molecules combinatorially with overly rigid constraints on potential rearrangements and poor time complexity or (b) by decoding lossy and nonphysical discretized spectra vectors. In this work, we introduce a new intermediate strategy for predicting mass spectra from molecules by treating mass spectra as sets of chemical formulae, which are themselves multisets of atoms. After first encoding an input molecular graph, we decode a set of chemical subformulae, each of which specify a predicted peak in the mass spectra, the intensities of which are predicted by a second model. Our key insight is to overcome the combinatorial possibilities for chemical subformulae by decoding the formula set using a prefix tree structure, atom-type by atom-type, representing a general method for ordered multiset decoding. We show promising empirical results on mass spectra prediction tasks.