 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMotifBench: A standardized protein design benchmark for motif-scaffolding problems
Feb 19, 2025The motif-scaffolding problem is a central task in computational protein design: Given the coordinates of atoms in a geometry chosen to confer a desired biochemical function (a motif), the task is to identify diverse protein structures (scaffolds) that include the motif and maintain its geometry. Significant recent progress on motif-scaffolding has been made due to computational evaluation with reliable protein structure prediction and fixed-backbone sequence design methods. However, significant variability in evaluation strategies across publications has hindered comparability of results, challenged reproducibility, and impeded robust progress. In response we introduce MotifBench, comprising (1) a precisely specified pipeline and evaluation metrics, (2) a collection of 30 benchmark problems, and (3) an implementation of this benchmark and leaderboard at github.com/blt2114/MotifBench. The MotifBench test cases are more difficult compared to earlier benchmarks, and include protein design problems for which solutions are known but on which, to the best of our knowledge, state-of-the-art methods fail to identify any solution.
Protein structure generation via folding diffusion
Sep 30, 2022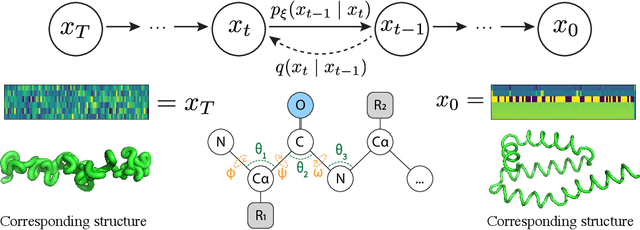

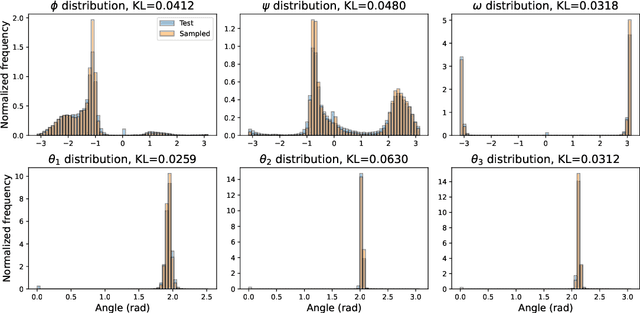
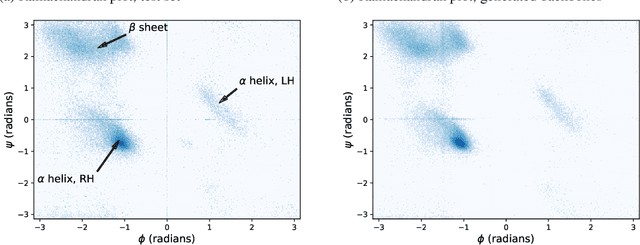
The ability to computationally generate novel yet physically foldable protein structures could lead to new biological discoveries and new treatments targeting yet incurable diseases. Despite recent advances in protein structure prediction, directly generating diverse, novel protein structures from neural networks remains difficult. In this work, we present a new diffusion-based generative model that designs protein backbone structures via a procedure that mirrors the native folding process. We describe protein backbone structure as a series of consecutive angles capturing the relative orientation of the constituent amino acid residues, and generate new structures by denoising from a random, unfolded state towards a stable folded structure. Not only does this mirror how proteins biologically twist into energetically favorable conformations, the inherent shift and rotational invariance of this representation crucially alleviates the need for complex equivariant networks. We train a denoising diffusion probabilistic model with a simple transformer backbone and demonstrate that our resulting model unconditionally generates highly realistic protein structures with complexity and structural patterns akin to those of naturally-occurring proteins. As a useful resource, we release the first open-source codebase and trained models for protein structure diffusion.
Exploring evolution-based & -free protein language models as protein function predictors
Jun 14, 2022
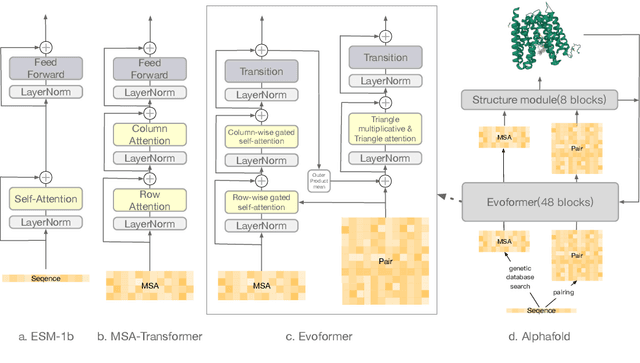
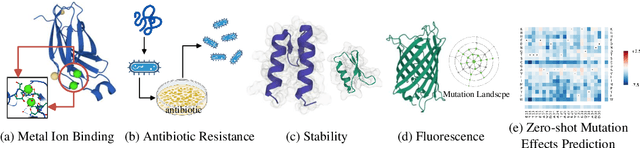

Large-scale Protein Language Models (PLMs) have improved performance in protein prediction tasks, ranging from 3D structure prediction to various function predictions. In particular, AlphaFold, a ground-breaking AI system, could potentially reshape structural biology. However, the utility of the PLM module in AlphaFold, Evoformer, has not been explored beyond structure prediction. In this paper, we investigate the representation ability of three popular PLMs: ESM-1b (single sequence), MSA-Transformer (multiple sequence alignment) and Evoformer (structural), with a special focus on Evoformer. Specifically, we aim to answer the following key questions: (i) Does the Evoformer trained as part of AlphaFold produce representations amenable to predicting protein function? (ii) If yes, can Evoformer replace ESM-1b and MSA-Transformer? (iii) How much do these PLMs rely on evolution-related protein data? In this regard, are they complementary to each other? We compare these models by empirical study along with new insights and conclusions. Finally, we release code and datasets for reproducibility.
Machine learning modeling of family wide enzyme-substrate specificity screens
Sep 08, 2021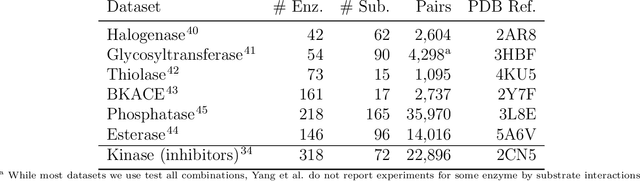
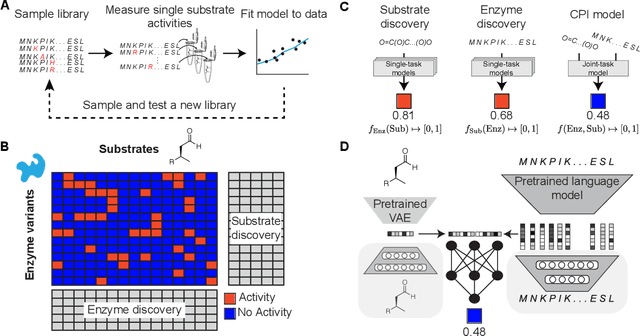
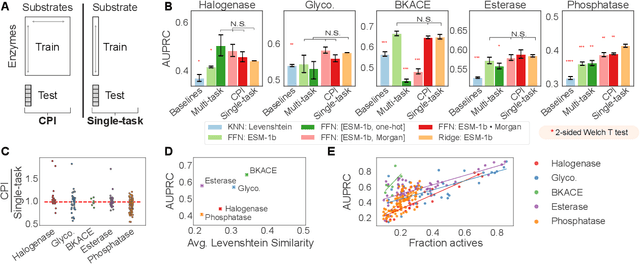
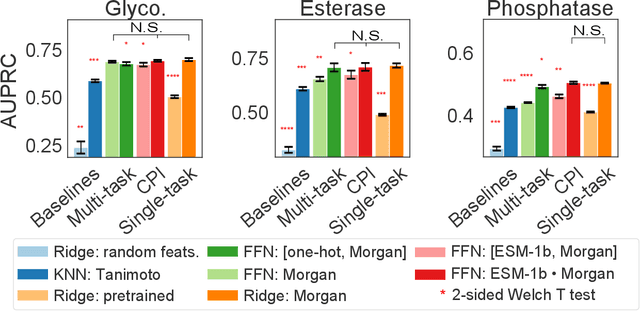
Biocatalysis is a promising approach to sustainably synthesize pharmaceuticals, complex natural products, and commodity chemicals at scale. However, the adoption of biocatalysis is limited by our ability to select enzymes that will catalyze their natural chemical transformation on non-natural substrates. While machine learning and in silico directed evolution are well-posed for this predictive modeling challenge, efforts to date have primarily aimed to increase activity against a single known substrate, rather than to identify enzymes capable of acting on new substrates of interest. To address this need, we curate 6 different high-quality enzyme family screens from the literature that each measure multiple enzymes against multiple substrates. We compare machine learning-based compound-protein interaction (CPI) modeling approaches from the literature used for predicting drug-target interactions. Surprisingly, comparing these interaction-based models against collections of independent (single task) enzyme-only or substrate-only models reveals that current CPI approaches are incapable of learning interactions between compounds and proteins in the current family level data regime. We further validate this observation by demonstrating that our no-interaction baseline can outperform CPI-based models from the literature used to guide the discovery of kinase inhibitors. Given the high performance of non-interaction based models, we introduce a new structure-based strategy for pooling residue representations across a protein sequence. Altogether, this work motivates a principled path forward in order to build and evaluate meaningful predictive models for biocatalysis and other drug discovery applications.
Adaptive machine learning for protein engineering
Jul 06, 2021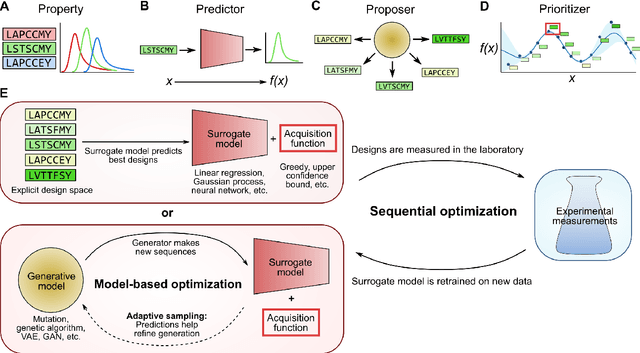

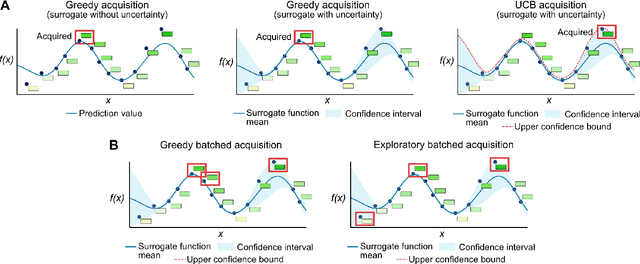
Machine-learning models that learn from data to predict how protein sequence encodes function are emerging as a useful protein engineering tool. However, when using these models to suggest new protein designs, one must deal with the vast combinatorial complexity of protein sequences. Here, we review how to use a sequence-to-function machine-learning surrogate model to select sequences for experimental measurement. First, we discuss how to select sequences through a single round of machine-learning optimization. Then, we discuss sequential optimization, where the goal is to discover optimized sequences and improve the model across multiple rounds of training, optimization, and experimental measurement.
Protein sequence design with deep generative models
Apr 09, 2021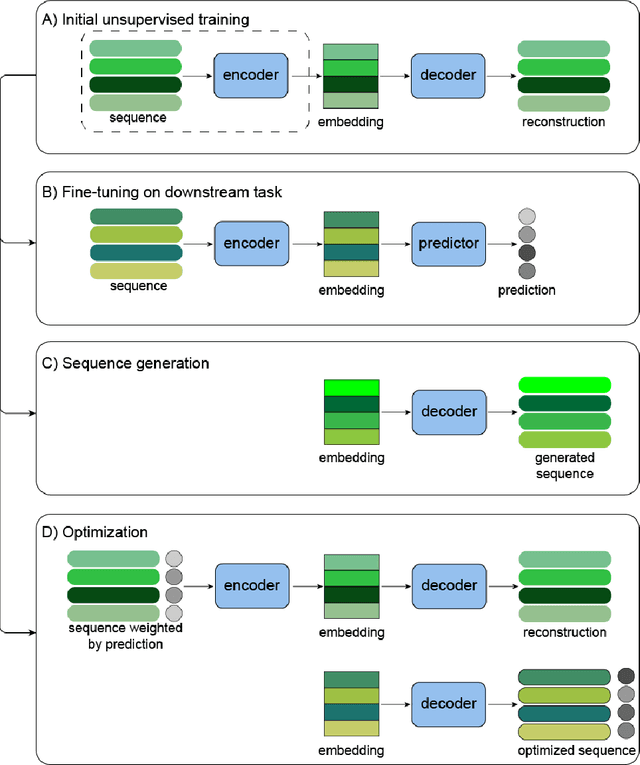
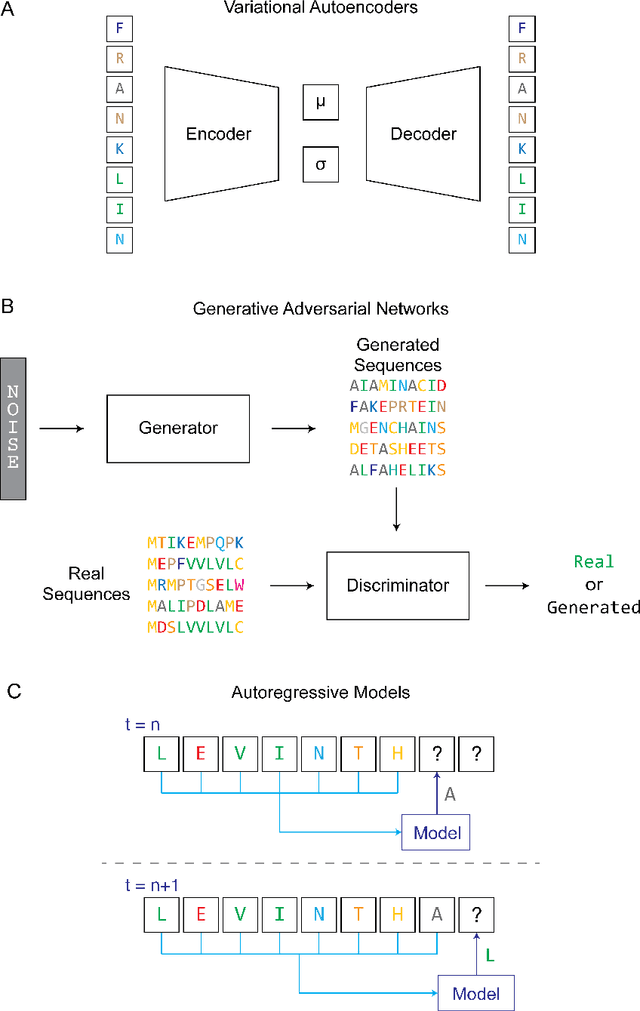
Protein engineering seeks to identify protein sequences with optimized properties. When guided by machine learning, protein sequence generation methods can draw on prior knowledge and experimental efforts to improve this process. In this review, we highlight recent applications of machine learning to generate protein sequences, focusing on the emerging field of deep generative methods.
Batched Stochastic Bayesian Optimization via Combinatorial Constraints Design
Apr 17, 2019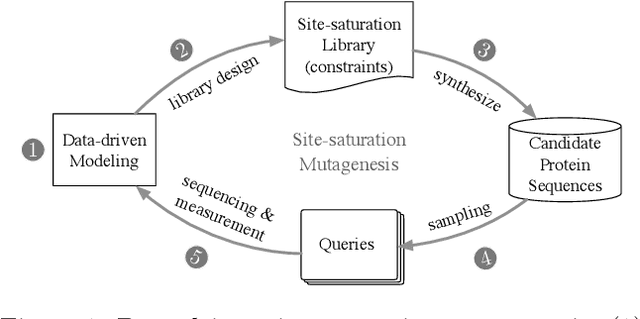
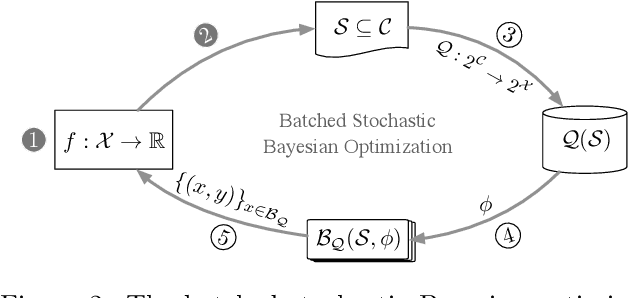
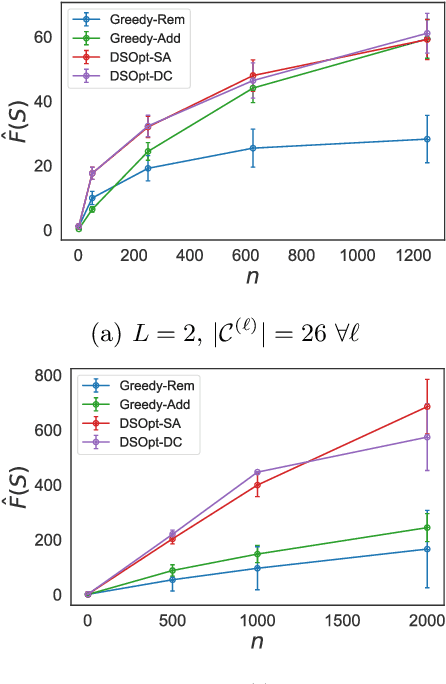
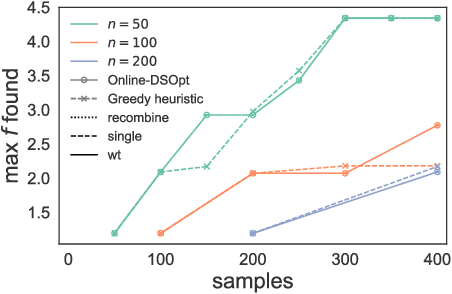
In many high-throughput experimental design settings, such as those common in biochemical engineering, batched queries are more cost effective than one-by-one sequential queries. Furthermore, it is often not possible to directly choose items to query. Instead, the experimenter specifies a set of constraints that generates a library of possible items, which are then selected stochastically. Motivated by these considerations, we investigate \emph{Batched Stochastic Bayesian Optimization} (BSBO), a novel Bayesian optimization scheme for choosing the constraints in order to guide exploration towards items with greater utility. We focus on \emph{site-saturation mutagenesis}, a prototypical setting of BSBO in biochemical engineering, and propose a natural objective function for this problem. Importantly, we show that our objective function can be efficiently decomposed as a difference of submodular functions (DS), which allows us to employ DS optimization tools to greedily identify sets of constraints that increase the likelihood of finding items with high utility. Our experimental results show that our algorithm outperforms common heuristics on both synthetic and two real protein datasets.