 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCausal integration of chemical structures improves representations of microscopy images for morphological profiling
Apr 13, 2025Recent advances in self-supervised deep learning have improved our ability to quantify cellular morphological changes in high-throughput microscopy screens, a process known as morphological profiling. However, most current methods only learn from images, despite many screens being inherently multimodal, as they involve both a chemical or genetic perturbation as well as an image-based readout. We hypothesized that incorporating chemical compound structure during self-supervised pre-training could improve learned representations of images in high-throughput microscopy screens. We introduce a representation learning framework, MICON (Molecular-Image Contrastive Learning), that models chemical compounds as treatments that induce counterfactual transformations of cell phenotypes. MICON significantly outperforms classical hand-crafted features such as CellProfiler and existing deep-learning-based representation learning methods in challenging evaluation settings where models must identify reproducible effects of drugs across independent replicates and data-generating centers. We demonstrate that incorporating chemical compound information into the learning process provides consistent improvements in our evaluation setting and that modeling compounds specifically as treatments in a causal framework outperforms approaches that directly align images and compounds in a single representation space. Our findings point to a new direction for representation learning in morphological profiling, suggesting that methods should explicitly account for the multimodal nature of microscopy screening data.
Continuous Time Evidential Distributions for Irregular Time Series
Jul 25, 2023



Prevalent in many real-world settings such as healthcare, irregular time series are challenging to formulate predictions from. It is difficult to infer the value of a feature at any given time when observations are sporadic, as it could take on a range of values depending on when it was last observed. To characterize this uncertainty we present EDICT, a strategy that learns an evidential distribution over irregular time series in continuous time. This distribution enables well-calibrated and flexible inference of partially observed features at any time of interest, while expanding uncertainty temporally for sparse, irregular observations. We demonstrate that EDICT attains competitive performance on challenging time series classification tasks and enabling uncertainty-guided inference when encountering noisy data.
Protein structure generation via folding diffusion
Sep 30, 2022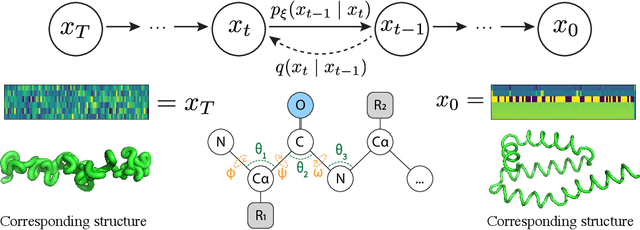

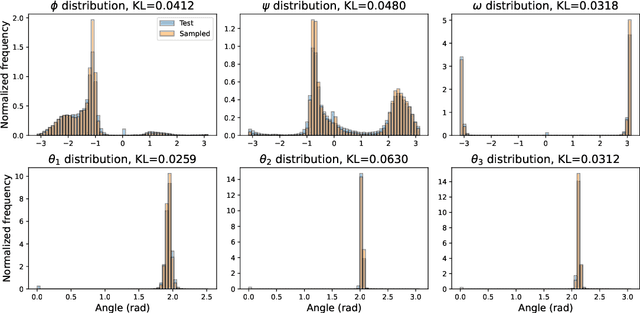
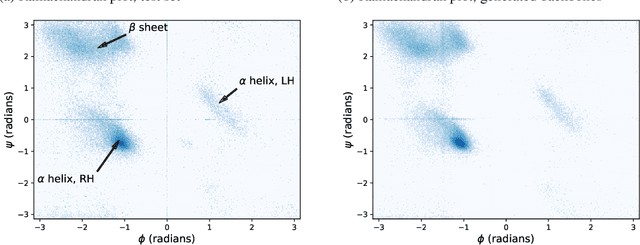
The ability to computationally generate novel yet physically foldable protein structures could lead to new biological discoveries and new treatments targeting yet incurable diseases. Despite recent advances in protein structure prediction, directly generating diverse, novel protein structures from neural networks remains difficult. In this work, we present a new diffusion-based generative model that designs protein backbone structures via a procedure that mirrors the native folding process. We describe protein backbone structure as a series of consecutive angles capturing the relative orientation of the constituent amino acid residues, and generate new structures by denoising from a random, unfolded state towards a stable folded structure. Not only does this mirror how proteins biologically twist into energetically favorable conformations, the inherent shift and rotational invariance of this representation crucially alleviates the need for complex equivariant networks. We train a denoising diffusion probabilistic model with a simple transformer backbone and demonstrate that our resulting model unconditionally generates highly realistic protein structures with complexity and structural patterns akin to those of naturally-occurring proteins. As a useful resource, we release the first open-source codebase and trained models for protein structure diffusion.