 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA probabilistic foundation model for crystal structure denoising, phase classification, and order parameters
Dec 21, 2025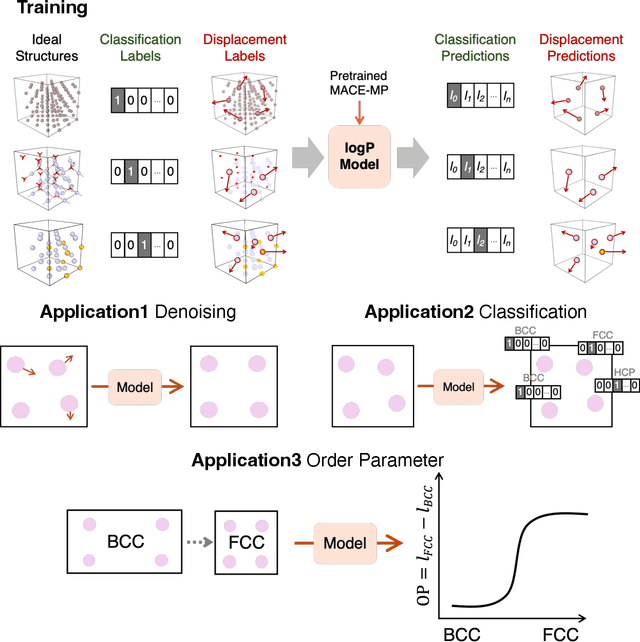
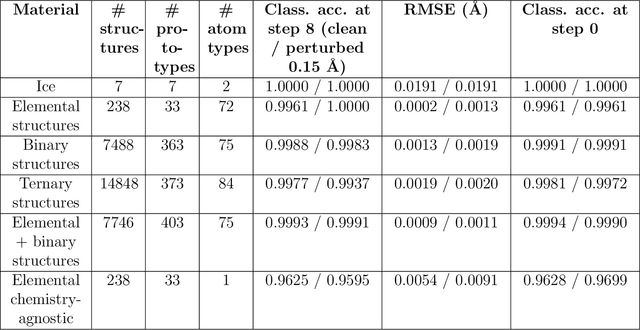
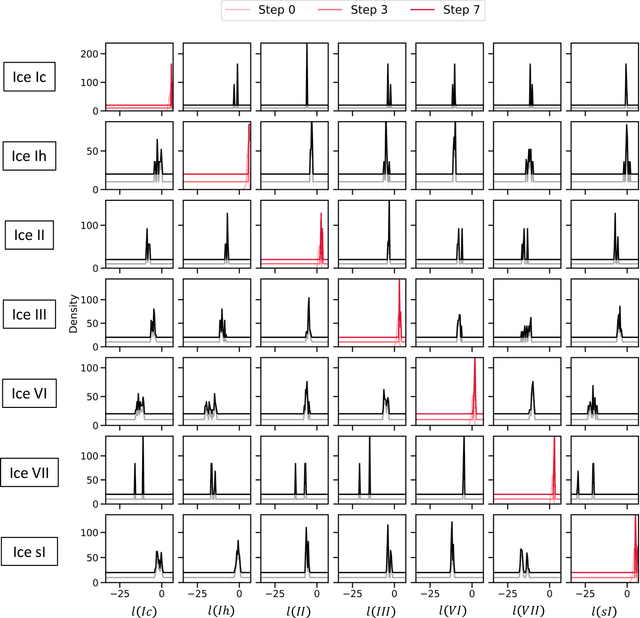
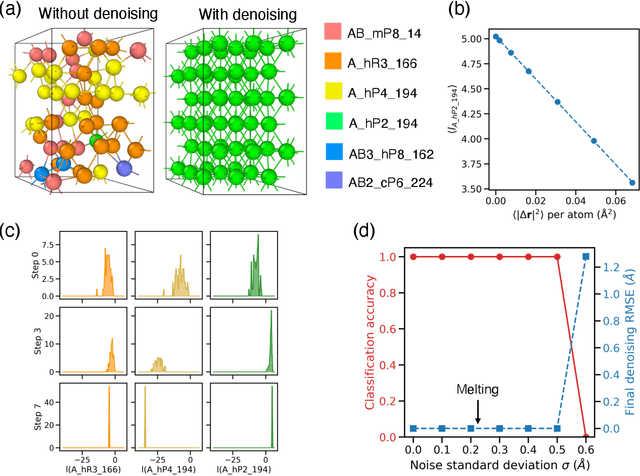
Atomistic simulations generate large volumes of noisy structural data, but extracting phase labels, order parameters (OPs), and defect information in a way that is universal, robust, and interpretable remains challenging. Existing tools such as PTM and CNA are restricted to a small set of hand-crafted lattices (e.g.\ FCC/BCC/HCP), degrade under strong thermal disorder or defects, and produce hard, template-based labels without per-atom probability or confidence scores. Here we introduce a log-probability foundation model that unifies denoising, phase classification, and OP extraction within a single probabilistic framework. We reuse the MACE-MP foundation interatomic potential on crystal structures mapped to AFLOW prototypes, training it to predict per-atom, per-phase logits $l$ and to aggregate them into a global log-density $\log \hat{P}_θ(\boldsymbol{r})$ whose gradient defines a conservative score field. Denoising corresponds to gradient ascent on this learned log-density, phase labels follow from $\arg\max_c l_{ac}$, and the $l$ values act as continuous, defect-sensitive and interpretable OPs quantifying the Euclidean distance to ideal phases. We demonstrate universality across hundreds of prototypes, robustness under strong thermal and defect-induced disorder, and accurate treatment of complex systems such as ice polymorphs, ice--water interfaces, and shock-compressed Ti.
Grand canonical generative diffusion model for crystalline phases and grain boundaries
Aug 28, 2024The diffusion model has emerged as a powerful tool for generating atomic structures for materials science. This work calls attention to the deficiency of current particle-based diffusion models, which represent atoms as a point cloud, in generating even the simplest ordered crystalline structures. The problem is attributed to particles being trapped in local minima during the score-driven simulated annealing of the diffusion process, similar to the physical process of force-driven simulated annealing. We develop a solution, the grand canonical diffusion model, which adopts an alternative voxel-based representation with continuous rather than fixed number of particles. The method is applied towards generation of several common crystalline phases as well as the technologically important and challenging problem of grain boundary structures.
Information theory unifies atomistic machine learning, uncertainty quantification, and materials thermodynamics
Apr 18, 2024An accurate description of information is relevant for a range of problems in atomistic modeling, such as sampling methods, detecting rare events, analyzing datasets, or performing uncertainty quantification (UQ) in machine learning (ML)-driven simulations. Although individual methods have been proposed for each of these tasks, they lack a common theoretical background integrating their solutions. Here, we introduce an information theoretical framework that unifies predictions of phase transformations, kinetic events, dataset optimality, and model-free UQ from atomistic simulations, thus bridging materials modeling, ML, and statistical mechanics. We first demonstrate that, for a proposed representation, the information entropy of a distribution of atom-centered environments is a surrogate value for thermodynamic entropy. Using molecular dynamics (MD) simulations, we show that information entropy differences from trajectories can be used to build phase diagrams, identify rare events, and recover classical theories of nucleation. Building on these results, we use this general concept of entropy to quantify information in datasets for ML interatomic potentials (IPs), informing compression, explaining trends in testing errors, and evaluating the efficiency of active learning strategies. Finally, we propose a model-free UQ method for MLIPs using information entropy, showing it reliably detects extrapolation regimes, scales to millions of atoms, and goes beyond model errors. This method is made available as the package QUESTS: Quick Uncertainty and Entropy via STructural Similarity, providing a new unifying theory for data-driven atomistic modeling and combining efforts in ML, first-principles thermodynamics, and simulations.
Score dynamics: scaling molecular dynamics with picosecond timesteps via conditional diffusion model
Oct 02, 2023We propose score dynamics (SD), a general framework for learning effective evolution operators for atomistic as well as coarse-grained dynamics from molecular-dynamics (MD) simulations. SD is centered around scores, or derivatives of the transition log-probability with respect to the dynamical degrees of freedom. The latter play the same role as force fields in MD but are used in denoising diffusion probability models to generate discrete transitions of the dynamical variables in an SD timestep, which can be orders of magnitude larger than a typical MD timestep. In this work, we construct graph neural network based score dynamics models of realistic molecular systems that are evolved with 1~ps timesteps. We demonstrate the efficacy of score dynamics with case studies of alanine dipeptide and short alkanes in aqueous solution. Both equilibrium predictions derived from the stationary distributions of the conditional probability and kinetic predictions for the transition rates and transition paths are in good agreement with MD at about 8-18 fold wall-clock speedup. Open challenges and possible future remedies to improve score dynamics are also discussed.
Score-based denoising for atomic structure identification
Dec 20, 2022We propose an accurate method for removing thermal vibrations that complicate the task of analyzing complex dynamics in atomistic simulation of condensed matter. Our method iteratively subtracts thermal noises or perturbations in atomic positions using a denoising score function trained on synthetically noised but otherwise perfect crystal lattices. The resulting denoised structures clearly reveal underlying crystal order while retaining disorder associated with crystal defects. Purely geometric, agnostic to interatomic potentials, and trained without inputs from explicit simulations, our denoiser can be applied to simulation data generated from vastly different interatomic interactions. Followed by a simple phase classification tool such as the Common Neighbor Analysis, the denoiser outperforms other existing methods and reaches perfect classification accuracy on a recently proposed benchmark dataset consisting of perturbed crystal structures (DC3). Demonstrated here in a wide variety of atomistic simulation contexts, the denoiser is general, robust, and readily extendable to delineate order from disorder in structurally and chemically complex materials.