 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBOOM: Benchmarking Out-Of-distribution Molecular Property Predictions of Machine Learning Models
May 03, 2025Advances in deep learning and generative modeling have driven interest in data-driven molecule discovery pipelines, whereby machine learning (ML) models are used to filter and design novel molecules without requiring prohibitively expensive first-principles simulations. Although the discovery of novel molecules that extend the boundaries of known chemistry requires accurate out-of-distribution (OOD) predictions, ML models often struggle to generalize OOD. Furthermore, there are currently no systematic benchmarks for molecular OOD prediction tasks. We present BOOM, $\boldsymbol{b}$enchmarks for $\boldsymbol{o}$ut-$\boldsymbol{o}$f-distribution $\boldsymbol{m}$olecular property predictions -- a benchmark study of property-based out-of-distribution models for common molecular property prediction models. We evaluate more than 140 combinations of models and property prediction tasks to benchmark deep learning models on their OOD performance. Overall, we do not find any existing models that achieve strong OOD generalization across all tasks: even the top performing model exhibited an average OOD error 3x larger than in-distribution. We find that deep learning models with high inductive bias can perform well on OOD tasks with simple, specific properties. Although chemical foundation models with transfer and in-context learning offer a promising solution for limited training data scenarios, we find that current foundation models do not show strong OOD extrapolation capabilities. We perform extensive ablation experiments to highlight how OOD performance is impacted by data generation, pre-training, hyperparameter optimization, model architecture, and molecular representation. We propose that developing ML models with strong OOD generalization is a new frontier challenge in chemical ML model development. This open-source benchmark will be made available on Github.
Grand canonical generative diffusion model for crystalline phases and grain boundaries
Aug 28, 2024The diffusion model has emerged as a powerful tool for generating atomic structures for materials science. This work calls attention to the deficiency of current particle-based diffusion models, which represent atoms as a point cloud, in generating even the simplest ordered crystalline structures. The problem is attributed to particles being trapped in local minima during the score-driven simulated annealing of the diffusion process, similar to the physical process of force-driven simulated annealing. We develop a solution, the grand canonical diffusion model, which adopts an alternative voxel-based representation with continuous rather than fixed number of particles. The method is applied towards generation of several common crystalline phases as well as the technologically important and challenging problem of grain boundary structures.
Cascading Blackout Severity Prediction with Statistically-Augmented Graph Neural Networks
Mar 22, 2024

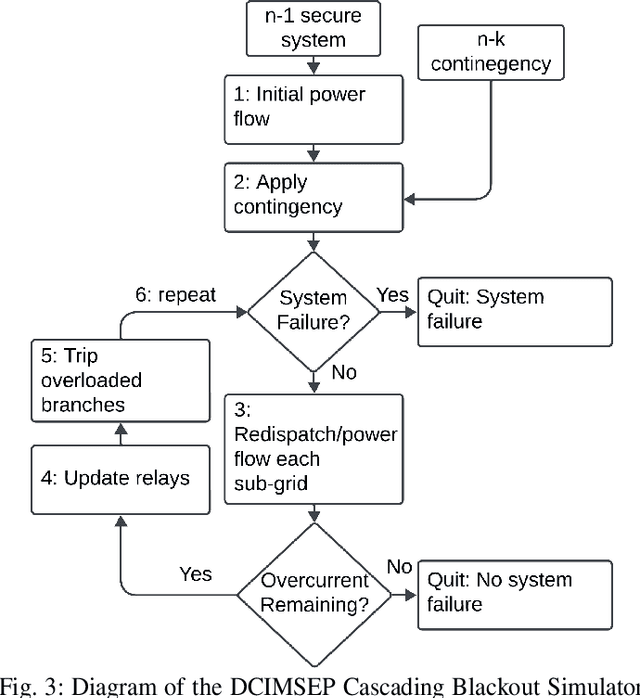

Higher variability in grid conditions, resulting from growing renewable penetration and increased incidence of extreme weather events, has increased the difficulty of screening for scenarios that may lead to catastrophic cascading failures. Traditional power-flow-based tools for assessing cascading blackout risk are too slow to properly explore the space of possible failures and load/generation patterns. We add to the growing literature of faster graph-neural-network (GNN)-based techniques, developing two novel techniques for the estimation of blackout magnitude from initial grid conditions. First we propose several methods for employing an initial classification step to filter out safe "non blackout" scenarios prior to magnitude estimation. Second, using insights from the statistical properties of cascading blackouts, we propose a method for facilitating non-local message passing in our GNN models. We validate these two approaches on a large simulated dataset, and show the potential of both to increase blackout size estimation performance.
Spectroscopy-Guided Discovery of Three-Dimensional Structures of Disordered Materials with Diffusion Models
Dec 09, 2023



The ability to rapidly develop materials with desired properties has a transformative impact on a broad range of emerging technologies. In this work, we introduce a new framework based on the diffusion model, a recent generative machine learning method to predict 3D structures of disordered materials from a target property. For demonstration, we apply the model to identify the atomic structures of amorphous carbons ($a$-C) as a representative material system from the target X-ray absorption near edge structure (XANES) spectra--a common experimental technique to probe atomic structures of materials. We show that conditional generation guided by XANES spectra reproduces key features of the target structures. Furthermore, we show that our model can steer the generative process to tailor atomic arrangements for a specific XANES spectrum. Finally, our generative model exhibits a remarkable scale-agnostic property, thereby enabling generation of realistic, large-scale structures through learning from a small-scale dataset (i.e., with small unit cells). Our work represents a significant stride in bridging the gap between materials characterization and atomic structure determination; in addition, it can be leveraged for materials discovery in exploring various material properties as targeted.
Score dynamics: scaling molecular dynamics with picosecond timesteps via conditional diffusion model
Oct 02, 2023We propose score dynamics (SD), a general framework for learning effective evolution operators for atomistic as well as coarse-grained dynamics from molecular-dynamics (MD) simulations. SD is centered around scores, or derivatives of the transition log-probability with respect to the dynamical degrees of freedom. The latter play the same role as force fields in MD but are used in denoising diffusion probability models to generate discrete transitions of the dynamical variables in an SD timestep, which can be orders of magnitude larger than a typical MD timestep. In this work, we construct graph neural network based score dynamics models of realistic molecular systems that are evolved with 1~ps timesteps. We demonstrate the efficacy of score dynamics with case studies of alanine dipeptide and short alkanes in aqueous solution. Both equilibrium predictions derived from the stationary distributions of the conditional probability and kinetic predictions for the transition rates and transition paths are in good agreement with MD at about 8-18 fold wall-clock speedup. Open challenges and possible future remedies to improve score dynamics are also discussed.
Score-based denoising for atomic structure identification
Dec 20, 2022We propose an accurate method for removing thermal vibrations that complicate the task of analyzing complex dynamics in atomistic simulation of condensed matter. Our method iteratively subtracts thermal noises or perturbations in atomic positions using a denoising score function trained on synthetically noised but otherwise perfect crystal lattices. The resulting denoised structures clearly reveal underlying crystal order while retaining disorder associated with crystal defects. Purely geometric, agnostic to interatomic potentials, and trained without inputs from explicit simulations, our denoiser can be applied to simulation data generated from vastly different interatomic interactions. Followed by a simple phase classification tool such as the Common Neighbor Analysis, the denoiser outperforms other existing methods and reaches perfect classification accuracy on a recently proposed benchmark dataset consisting of perturbed crystal structures (DC3). Demonstrated here in a wide variety of atomistic simulation contexts, the denoiser is general, robust, and readily extendable to delineate order from disorder in structurally and chemically complex materials.
Challenges and approaches to privacy preserving post-click conversion prediction
Jan 29, 2022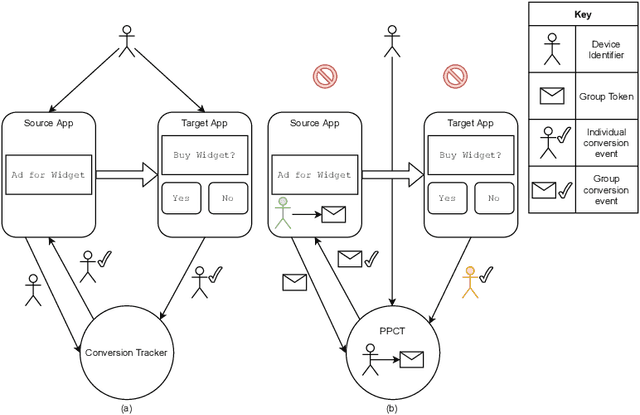
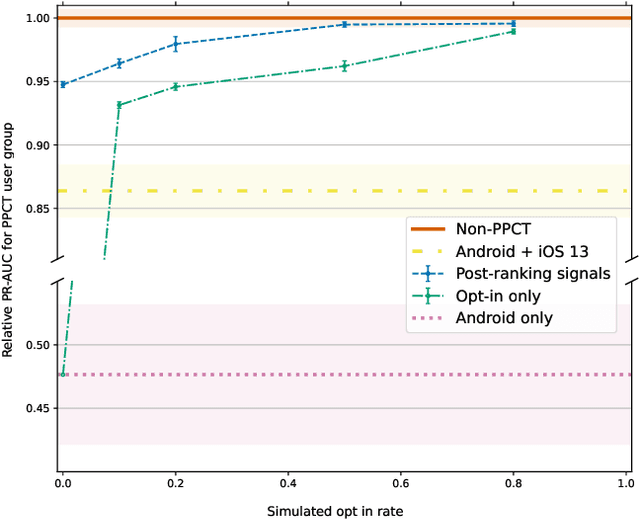
Online advertising has typically been more personalized than offline advertising, through the use of machine learning models and real-time auctions for ad targeting. One specific task, predicting the likelihood of conversion (i.e.\ the probability a user will purchase the advertised product), is crucial to the advertising ecosystem for both targeting and pricing ads. Currently, these models are often trained by observing individual user behavior, but, increasingly, regulatory and technical constraints are requiring privacy-preserving approaches. For example, major platforms are moving to restrict tracking individual user events across multiple applications, and governments around the world have shown steadily more interest in regulating the use of personal data. Instead of receiving data about individual user behavior, advertisers may receive privacy-preserving feedback, such as the number of installs of an advertised app that resulted from a group of users. In this paper we outline the recent privacy-related changes in the online advertising ecosystem from a machine learning perspective. We provide an overview of the challenges and constraints when learning conversion models in this setting. We introduce a novel approach for training these models that makes use of post-ranking signals. We show using offline experiments on real world data that it outperforms a model relying on opt-in data alone, and significantly reduces model degradation when no individual labels are available. Finally, we discuss future directions for research in this evolving area.
Efficient, Interpretable Atomistic Graph Neural Network Representation for Angle-dependent Properties and its Application to Optical Spectroscopy Prediction
Sep 23, 2021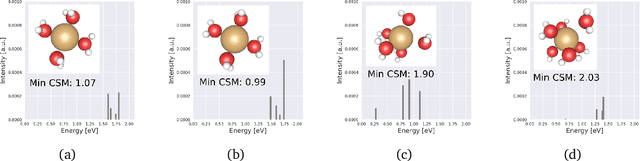


Graph neural networks (GNNs) are attractive for learning properties of atomic structures thanks to their intuitive, physically informed graph encoding of atoms and bonds. However, conventional GNN encodings do not account for angular information, which is critical for describing complex atomic arrangements in disordered materials, interfaces, and molecular distortions. In this work, we extend the recently proposed ALIGNN encoding, which incorporates bond angles, to also include dihedral angles (ALIGNN-d), and we apply the model to capture the structures of aqua copper complexes for spectroscopy prediction. This simple extension is shown to lead to a memory-efficient graph representation capable of capturing the full geometric information of atomic structures. Specifically, the ALIGNN-d encoding is a sparse yet equally expressive representation compared to the dense, maximally-connected graph, in which all bonds are encoded. We also explore model interpretability based on ALIGNN-d by elucidating the relative contributions of individual structural components to the optical response of the copper complexes. Lastly, we briefly discuss future developments to validate the computational efficiency and to extend the interpretability of ALIGNN-d.
Microstructure Generation via Generative Adversarial Network for Heterogeneous, Topologically Complex 3D Materials
Jun 22, 2020
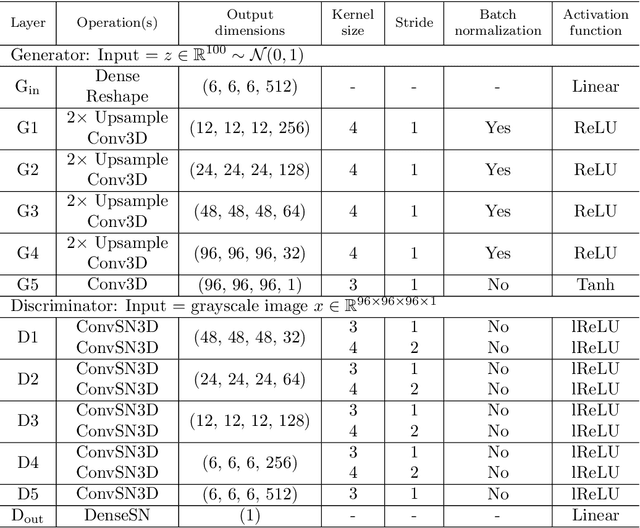
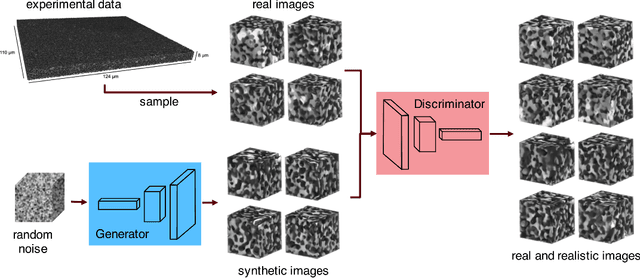

Using a large-scale, experimentally captured 3D microstructure dataset, we implement the generative adversarial network (GAN) framework to learn and generate 3D microstructures of solid oxide fuel cell electrodes. The generated microstructures are visually, statistically, and topologically realistic, with distributions of microstructural parameters, including volume fraction, particle size, surface area, tortuosity, and triple phase boundary density, being highly similar to those of the original microstructure. These results are compared and contrasted with those from an established, grain-based generation algorithm (DREAM.3D). Importantly, simulations of electrochemical performance, using a locally resolved finite element model, demonstrate that the GAN generated microstructures closely match the performance distribution of the original, while DREAM.3D leads to significant differences. The ability of the generative machine learning model to recreate microstructures with high fidelity suggests that the essence of complex microstructures may be captured and represented in a compact and manipulatable form.