 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeActive Learning Enables Extrapolation in Molecular Generative Models
Jan 03, 2025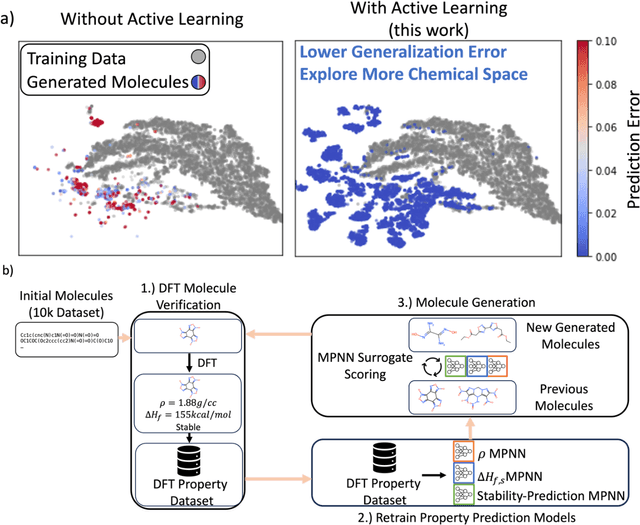
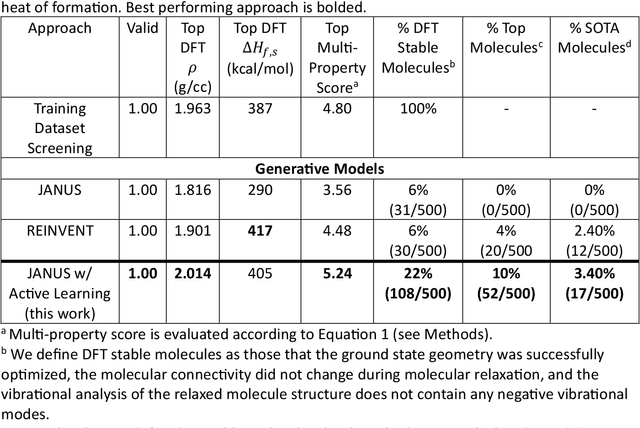
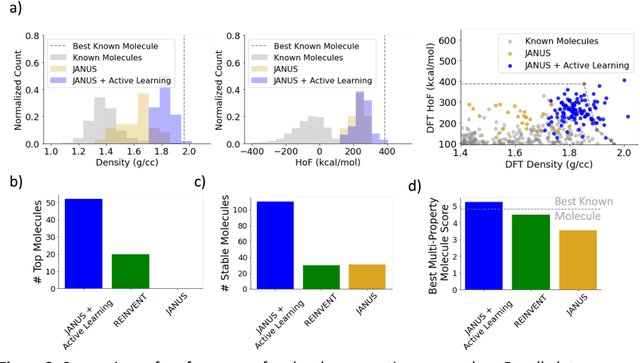
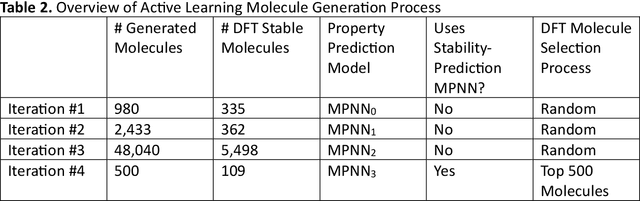
Although generative models hold promise for discovering molecules with optimized desired properties, they often fail to suggest synthesizable molecules that improve upon the known molecules seen in training. We find that a key limitation is not in the molecule generation process itself, but in the poor generalization capabilities of molecular property predictors. We tackle this challenge by creating an active-learning, closed-loop molecule generation pipeline, whereby molecular generative models are iteratively refined on feedback from quantum chemical simulations to improve generalization to new chemical space. Compared against other generative model approaches, only our active learning approach generates molecules with properties that extrapolate beyond the training data (reaching up to 0.44 standard deviations beyond the training data range) and out-of-distribution molecule classification accuracy is improved by 79%. By conditioning molecular generation on thermodynamic stability data from the active-learning loop, the proportion of stable molecules generated is 3.5x higher than the next-best model.
Efficient, Interpretable Atomistic Graph Neural Network Representation for Angle-dependent Properties and its Application to Optical Spectroscopy Prediction
Sep 23, 2021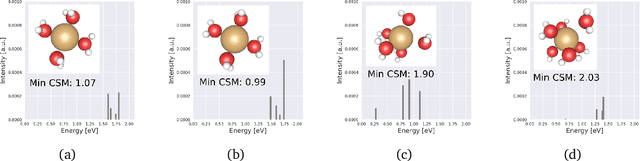

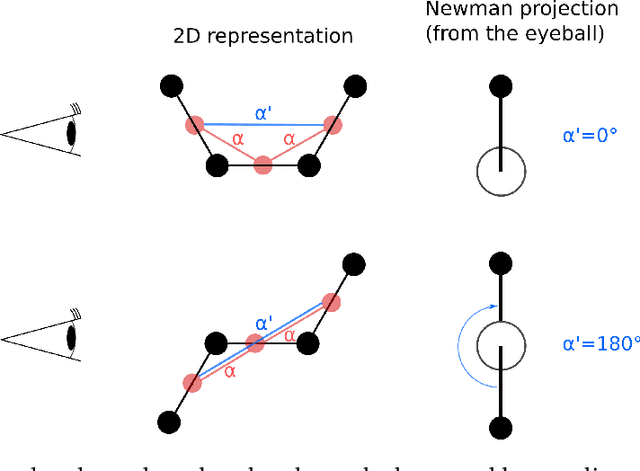

Graph neural networks (GNNs) are attractive for learning properties of atomic structures thanks to their intuitive, physically informed graph encoding of atoms and bonds. However, conventional GNN encodings do not account for angular information, which is critical for describing complex atomic arrangements in disordered materials, interfaces, and molecular distortions. In this work, we extend the recently proposed ALIGNN encoding, which incorporates bond angles, to also include dihedral angles (ALIGNN-d), and we apply the model to capture the structures of aqua copper complexes for spectroscopy prediction. This simple extension is shown to lead to a memory-efficient graph representation capable of capturing the full geometric information of atomic structures. Specifically, the ALIGNN-d encoding is a sparse yet equally expressive representation compared to the dense, maximally-connected graph, in which all bonds are encoded. We also explore model interpretability based on ALIGNN-d by elucidating the relative contributions of individual structural components to the optical response of the copper complexes. Lastly, we briefly discuss future developments to validate the computational efficiency and to extend the interpretability of ALIGNN-d.