 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine Learning-driven Multiscale MD Workflows: The Mini-MuMMI Experience
Jul 10, 2025Computational models have become one of the prevalent methods to model complex phenomena. To accurately model complex interactions, such as detailed biomolecular interactions, scientists often rely on multiscale models comprised of several internal models operating at difference scales, ranging from microscopic to macroscopic length and time scales. Bridging the gap between different time and length scales has historically been challenging but the advent of newer machine learning (ML) approaches has shown promise for tackling that task. Multiscale models require massive amounts of computational power and a powerful workflow management system. Orchestrating ML-driven multiscale studies on parallel systems with thousands of nodes is challenging, the workflow must schedule, allocate and control thousands of simulations operating at different scales. Here, we discuss the massively parallel Multiscale Machine-Learned Modeling Infrastructure (MuMMI), a multiscale workflow management infrastructure, that can orchestrate thousands of molecular dynamics (MD) simulations operating at different timescales, spanning from millisecond to nanosecond. More specifically, we introduce a novel version of MuMMI called "mini-MuMMI". Mini-MuMMI is a curated version of MuMMI designed to run on modest HPC systems or even laptops whereas MuMMI requires larger HPC systems. We demonstrate mini-MuMMI utility by exploring RAS-RAF membrane interactions and discuss the different challenges behind the generalization of multiscale workflows and how mini-MuMMI can be leveraged to target a broader range of applications outside of MD and RAS-RAF interactions.
Spectroscopy-Guided Discovery of Three-Dimensional Structures of Disordered Materials with Diffusion Models
Dec 09, 2023



The ability to rapidly develop materials with desired properties has a transformative impact on a broad range of emerging technologies. In this work, we introduce a new framework based on the diffusion model, a recent generative machine learning method to predict 3D structures of disordered materials from a target property. For demonstration, we apply the model to identify the atomic structures of amorphous carbons ($a$-C) as a representative material system from the target X-ray absorption near edge structure (XANES) spectra--a common experimental technique to probe atomic structures of materials. We show that conditional generation guided by XANES spectra reproduces key features of the target structures. Furthermore, we show that our model can steer the generative process to tailor atomic arrangements for a specific XANES spectrum. Finally, our generative model exhibits a remarkable scale-agnostic property, thereby enabling generation of realistic, large-scale structures through learning from a small-scale dataset (i.e., with small unit cells). Our work represents a significant stride in bridging the gap between materials characterization and atomic structure determination; in addition, it can be leveraged for materials discovery in exploring various material properties as targeted.
Identifying Orientation-specific Lipid-protein Fingerprints using Deep Learning
Jul 14, 2022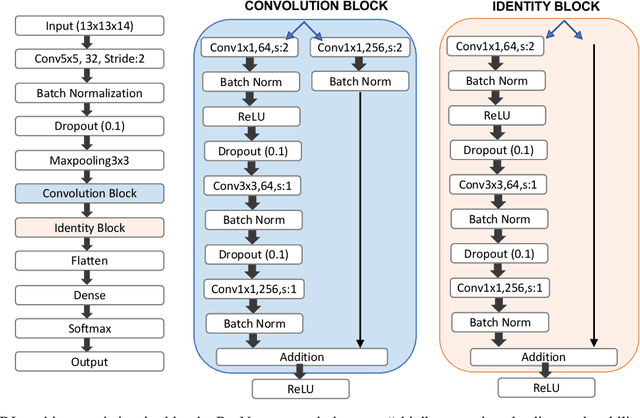
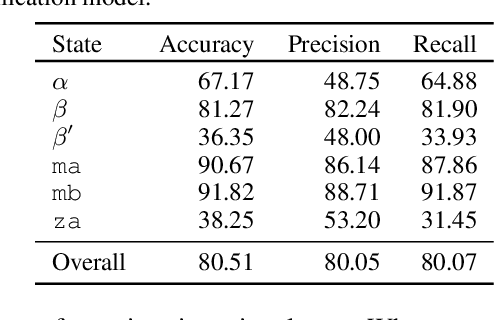
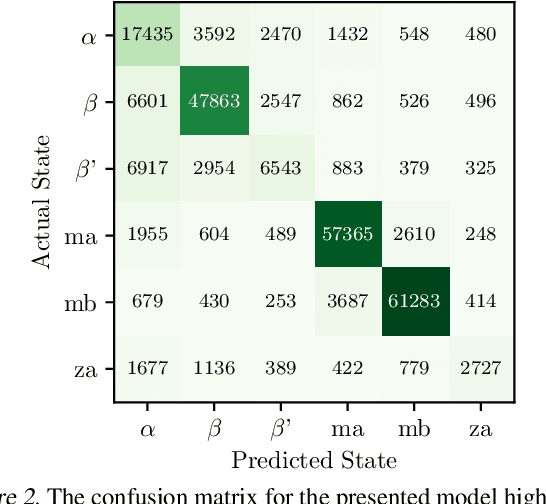
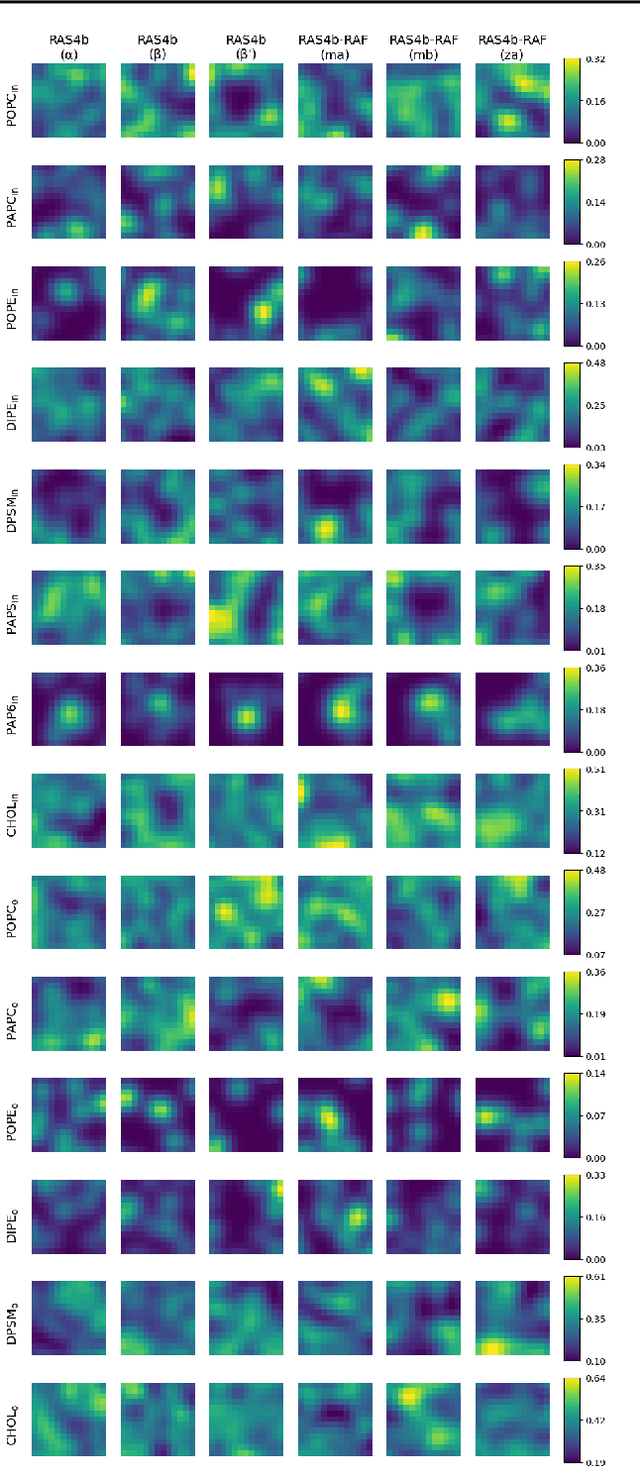
Improved understanding of the relation between the behavior of RAS and RAF proteins and the local lipid environment in the cell membrane is critical for getting insights into the mechanisms underlying cancer formation. In this work, we employ deep learning (DL) to learn this relationship by predicting protein orientational states of RAS and RAS-RAF protein complexes with respect to the lipid membrane based on the lipid densities around the protein domains from coarse-grained (CG) molecular dynamics (MD) simulations. Our DL model can predict six protein states with an overall accuracy of over 80%. The findings of this work offer new insights into how the proteins modulate the lipid environment, which in turn may assist designing novel therapies to regulate such interactions in the mechanisms associated with cancer development.
Emerging Patterns in the Continuum Representation of Protein-Lipid Fingerprints
Jul 09, 2022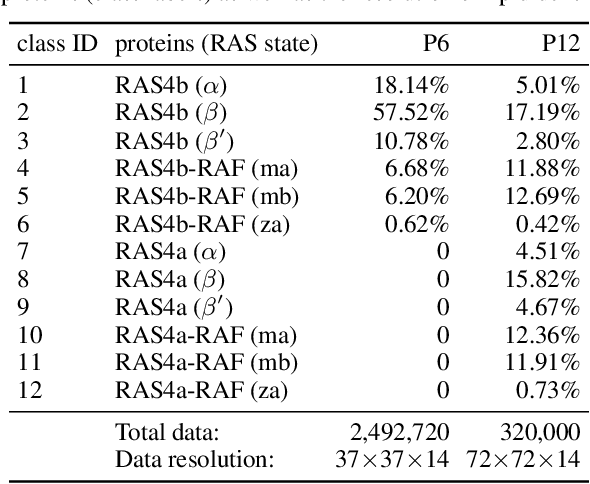
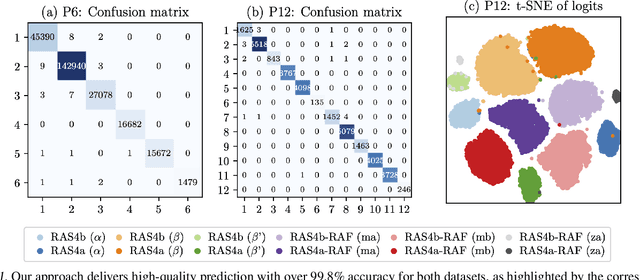
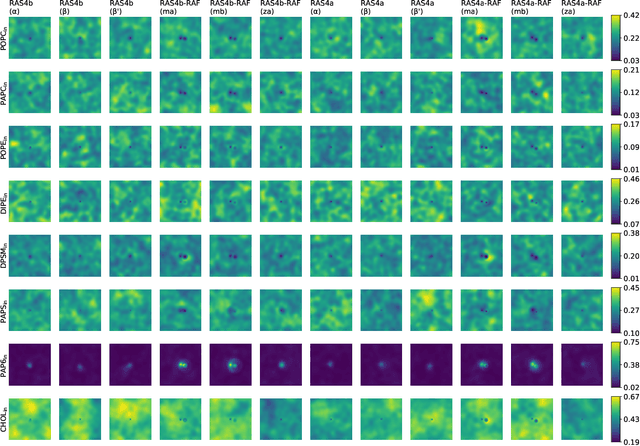
Capturing intricate biological phenomena often requires multiscale modeling where coarse and inexpensive models are developed using limited components of expensive and high-fidelity models. Here, we consider such a multiscale framework in the context of cancer biology and address the challenge of evaluating the descriptive capabilities of a continuum model developed using 1-dimensional statistics from a molecular dynamics model. Using deep learning, we develop a highly predictive classification model that identifies complex and emergent behavior from the continuum model. With over 99.9% accuracy demonstrated for two simulations, our approach confirms the existence of protein-specific "lipid fingerprints", i.e. spatial rearrangements of lipids in response to proteins of interest. Through this demonstration, our model also provides external validation of the continuum model, affirms the value of such multiscale modeling, and can foster new insights through further analysis of these fingerprints.