 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBOOM: Benchmarking Out-Of-distribution Molecular Property Predictions of Machine Learning Models
May 03, 2025Advances in deep learning and generative modeling have driven interest in data-driven molecule discovery pipelines, whereby machine learning (ML) models are used to filter and design novel molecules without requiring prohibitively expensive first-principles simulations. Although the discovery of novel molecules that extend the boundaries of known chemistry requires accurate out-of-distribution (OOD) predictions, ML models often struggle to generalize OOD. Furthermore, there are currently no systematic benchmarks for molecular OOD prediction tasks. We present BOOM, $\boldsymbol{b}$enchmarks for $\boldsymbol{o}$ut-$\boldsymbol{o}$f-distribution $\boldsymbol{m}$olecular property predictions -- a benchmark study of property-based out-of-distribution models for common molecular property prediction models. We evaluate more than 140 combinations of models and property prediction tasks to benchmark deep learning models on their OOD performance. Overall, we do not find any existing models that achieve strong OOD generalization across all tasks: even the top performing model exhibited an average OOD error 3x larger than in-distribution. We find that deep learning models with high inductive bias can perform well on OOD tasks with simple, specific properties. Although chemical foundation models with transfer and in-context learning offer a promising solution for limited training data scenarios, we find that current foundation models do not show strong OOD extrapolation capabilities. We perform extensive ablation experiments to highlight how OOD performance is impacted by data generation, pre-training, hyperparameter optimization, model architecture, and molecular representation. We propose that developing ML models with strong OOD generalization is a new frontier challenge in chemical ML model development. This open-source benchmark will be made available on Github.
PtychoFormer: A Transformer-based Model for Ptychographic Phase Retrieval
Oct 22, 2024



Ptychography is a computational method of microscopy that recovers high-resolution transmission images of samples from a series of diffraction patterns. While conventional phase retrieval algorithms can iteratively recover the images, they require oversampled diffraction patterns, incur significant computational costs, and struggle to recover the absolute phase of the sample's transmission function. Deep learning algorithms for ptychography are a promising approach to resolving the limitations of iterative algorithms. We present PtychoFormer, a hierarchical transformer-based model for data-driven single-shot ptychographic phase retrieval. PtychoFormer processes subsets of diffraction patterns, generating local inferences that are seamlessly stitched together to produce a high-quality reconstruction. Our model exhibits tolerance to sparsely scanned diffraction patterns and achieves up to 3600 times faster imaging speed than the extended ptychographic iterative engine (ePIE). We also propose the extended-PtychoFormer (ePF), a hybrid approach that combines the benefits of PtychoFormer with the ePIE. ePF minimizes global phase shifts and significantly enhances reconstruction quality, achieving state-of-the-art phase retrieval in ptychography.
Distributed Reinforcement Learning for Molecular Design: Antioxidant case
Dec 03, 2023Deep reinforcement learning has successfully been applied for molecular discovery as shown by the Molecule Deep Q-network (MolDQN) algorithm. This algorithm has challenges when applied to optimizing new molecules: training such a model is limited in terms of scalability to larger datasets and the trained model cannot be generalized to different molecules in the same dataset. In this paper, a distributed reinforcement learning algorithm for antioxidants, called DA-MolDQN is proposed to address these problems. State-of-the-art bond dissociation energy (BDE) and ionization potential (IP) predictors are integrated into DA-MolDQN, which are critical chemical properties while optimizing antioxidants. Training time is reduced by algorithmic improvements for molecular modifications. The algorithm is distributed, scalable for up to 512 molecules, and generalizes the model to a diverse set of molecules. The proposed models are trained with a proprietary antioxidant dataset. The results have been reproduced with both proprietary and public datasets. The proposed molecules have been validated with DFT simulations and a subset of them confirmed in public "unseen" datasets. In summary, DA-MolDQN is up to 100x faster than previous algorithms and can discover new optimized molecules from proprietary and public antioxidants.
STRIDE: Structure-guided Generation for Inverse Design of Molecules
Nov 06, 2023Machine learning and especially deep learning has had an increasing impact on molecule and materials design. In particular, given the growing access to an abundance of high-quality small molecule data for generative modeling for drug design, results for drug discovery have been promising. However, for many important classes of materials such as catalysts, antioxidants, and metal-organic frameworks, such large datasets are not available. Such families of molecules with limited samples and structural similarities are especially prevalent for industrial applications. As is well-known, retraining and even fine-tuning are challenging on such small datasets. Novel, practically applicable molecules are most often derivatives of well-known molecules, suggesting approaches to addressing data scarcity. To address this problem, we introduce $\textbf{STRIDE}$, a generative molecule workflow that generates novel molecules with an unconditional generative model guided by known molecules without any retraining. We generate molecules outside of the training data from a highly specialized set of antioxidant molecules. Our generated molecules have on average 21.7% lower synthetic accessibility scores and also reduce ionization potential by 5.9% of generated molecules via guiding.
ParticleGrid: Enabling Deep Learning using 3D Representation of Materials
Nov 15, 2022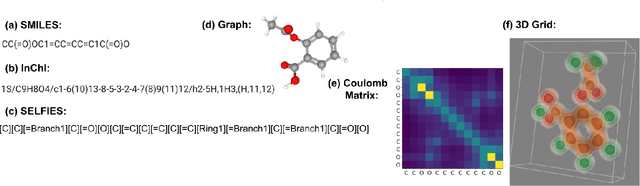
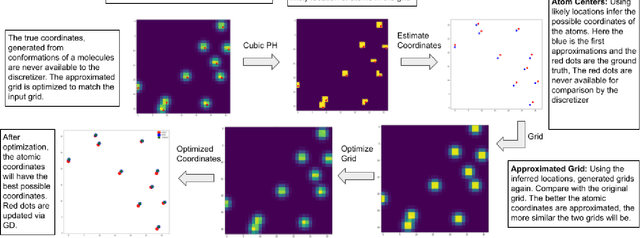
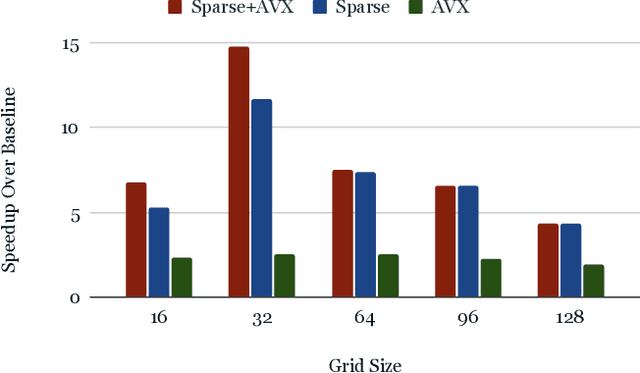
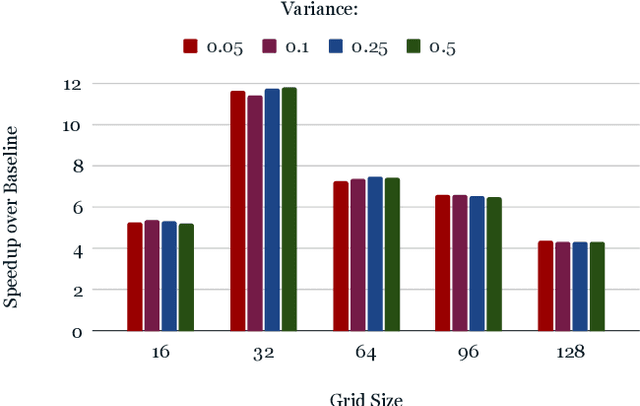
From AlexNet to Inception, autoencoders to diffusion models, the development of novel and powerful deep learning models and learning algorithms has proceeded at breakneck speeds. In part, we believe that rapid iteration of model architecture and learning techniques by a large community of researchers over a common representation of the underlying entities has resulted in transferable deep learning knowledge. As a result, model scale, accuracy, fidelity, and compute performance have dramatically increased in computer vision and natural language processing. On the other hand, the lack of a common representation for chemical structure has hampered similar progress. To enable transferable deep learning, we identify the need for a robust 3-dimensional representation of materials such as molecules and crystals. The goal is to enable both materials property prediction and materials generation with 3D structures. While computationally costly, such representations can model a large set of chemical structures. We propose $\textit{ParticleGrid}$, a SIMD-optimized library for 3D structures, that is designed for deep learning applications and to seamlessly integrate with deep learning frameworks. Our highly optimized grid generation allows for generating grids on the fly on the CPU, reducing storage and GPU compute and memory requirements. We show the efficacy of 3D grids generated via $\textit{ParticleGrid}$ and accurately predict molecular energy properties using a 3D convolutional neural network. Our model is able to get 0.006 mean square error and nearly match the values calculated using computationally costly density functional theory at a fraction of the time.
Graph Neural Network for Metal Organic Framework Potential Energy Approximation
Oct 29, 2020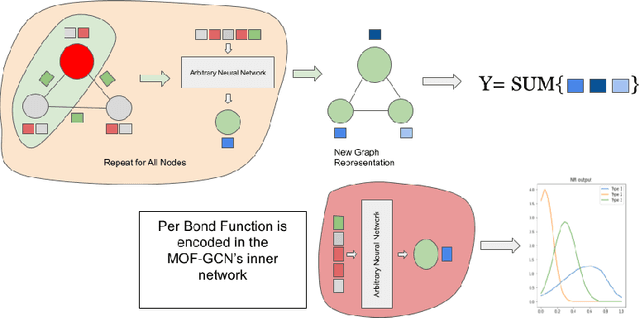
Metal-organic frameworks (MOFs) are nanoporous compounds composed of metal ions and organic linkers. MOFs play an important role in industrial applications such as gas separation, gas purification, and electrolytic catalysis. Important MOF properties such as potential energy are currently computed via techniques such as density functional theory (DFT). Although DFT provides accurate results, it is computationally costly. We propose a machine learning approach for estimating the potential energy of candidate MOFs, decomposing it into separate pair-wise atomic interactions using a graph neural network. Such a technique will allow high-throughput screening of candidates MOFs. We also generate a database of 50,000 spatial configurations and high-quality potential energy values using DFT.
Towards Run Time Estimation of the Gaussian Chemistry Code for SEAGrid Science Gateway
Jun 07, 2019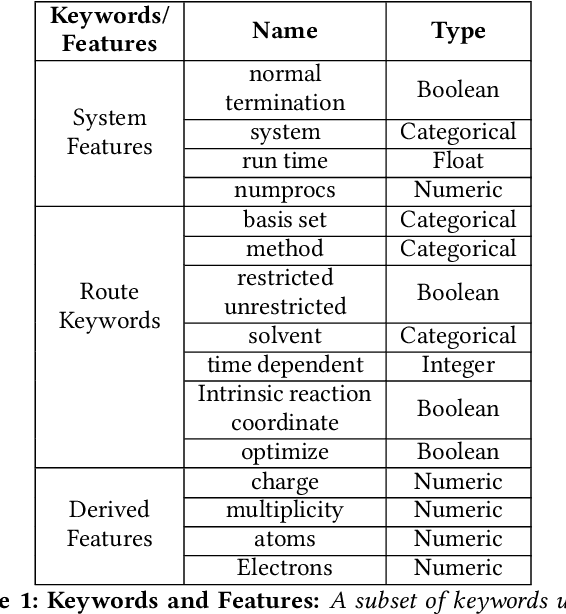
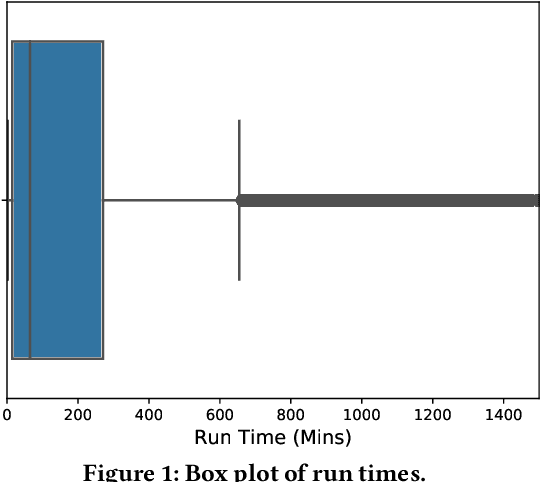
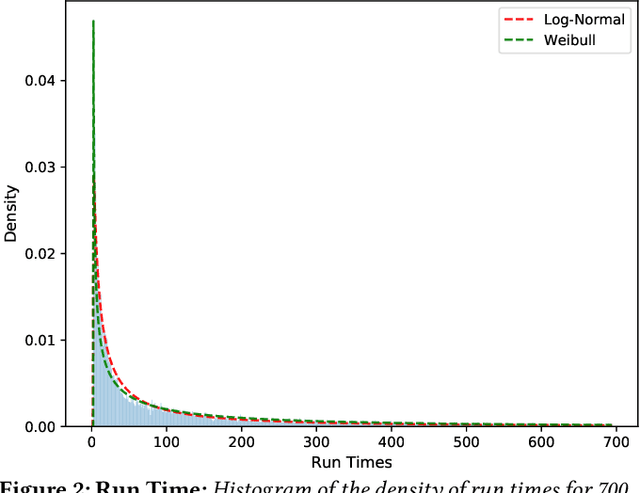
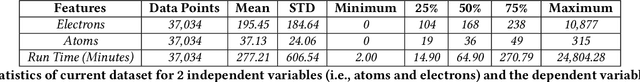
Accurate estimation of the run time of computational codes has a number of significant advantages for scientific computing. It is required information for optimal resource allocation, improving turnaround times and utilization of science gateways. Furthermore, it allows users to better plan and schedule their research, streamlining workflows and improving the overall productivity of cyberinfrastructure. Predicting run time is challenging, however. The inputs to scientific codes can be complex and high dimensional. Their relationship to the run time may be highly non-linear, and, in the most general case is completely arbitrary and thus unpredictable (i.e., simply a random mapping from inputs to run time). Most codes are not so arbitrary, however, and there has been significant prior research on predicting the run time of applications and workloads. Such predictions are generally application-specific, however. In this paper, we focus on the Gaussian computational chemistry code. We characterize a data set of runs from the SEAGrid science gateway with a number of different studies. We also explore a number of different potential regression methods and present promising future directions.